











- DAZ.online
- News
- Spektrum
- Studie stoppte trotz ...
ANSM Prüfbericht zu BIAL / BIotrial
Studie stoppte trotz Krankenhauseinweisung des ersten Probanden nicht
Rennes / Paris - 28.01.2016, 09:30 Uhr
Das Krankenhaus in Rennes: Nach dem ersten Zwischenbericht der französischen Arzneimittelbehörde werden weitere Probleme bei der Studie von Bial und Biotrial deutlich. (Foto: dpa)
Zwei Wochen nach dem tödlichen Zwischenfall in einer klinischen Studie der Firma Bial hat die französische Arzneimittelbehörde einen ersten Zwischenbericht veröffentlicht. Dieser zeigt, dass noch am Tag, nachdem der später verstorbene Proband in ein Krankenhaus eingeliefert worden war, der Wirkstoff an weitere Probanden der Studie gegeben wurde.
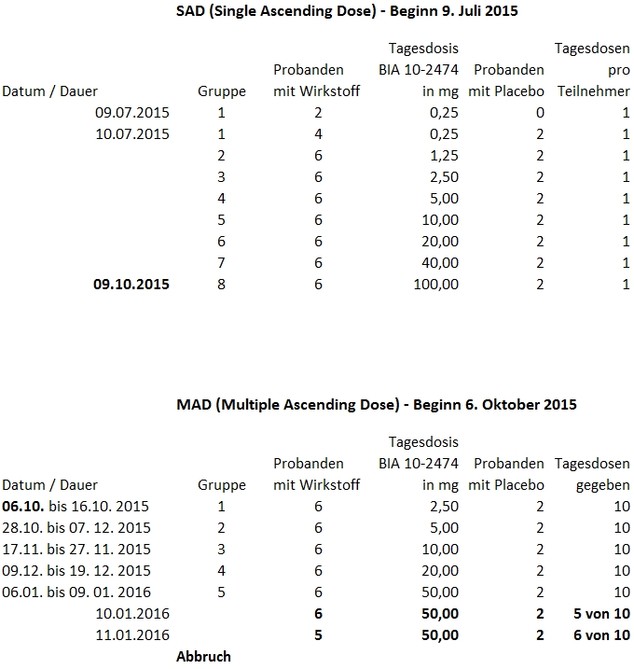
Der erste Bericht der französischen Arzneimittelbehörde ANSM zu den Ereignissen um die klinische Studie von BIA 10-2474, durchgeführt durch das französische Prüflabor Biotrial in Rennes, wurde am späten Mittwochnachmittag veröffentlicht. Erstmals werden wichtige Details klar: Der Zwischenbericht listet auf, welche Probanden wann welche Dosis von BIA 10-2474 erhalten haben. Und er deckt weitere Mängel auf.
So enthält er ein folgenschweres Detail zum tragischen Zwischenfall: Nachdem der später verstorbene Proband am 10. Januar in die Notaufnahme der Uniklinik eingewiesen wurde, wurde die Studie an den anderen Probanden seiner Gruppe am folgenden Tag weitergeführt.
Die französische Gesundheitsministerin Marisol Touraine hatte auf einer Pressekonferenz erklärt, die Studie sei „am Tag darauf“ gestoppt worden. Doch am Morgen bekamen die restlichen fünf Patienten zuerst noch eine Dosis BIA 10-2474, zwei Probanden ein Placebo. Bei drei der Probanden wurden später schwere Nebenwirkungen festgestellt.
Marisol Touraine, die französische Gesundheitsministerin, und Gilles Edan, Leiter der Neurologie am Uniklinikum Rennes, während einer Pressekonferenz. (Foto: dpa)
Das ist laut den französischen Prüfern passiert:
Wie das Studienprotokoll vorsah, konnten die einzelnen Studienteile überlappend durchgeführt werden. Und so begann der Testteil „Multiple Ascending Dose“ (MAD), bei dem es später zu den Zwischenfällen kam, tatsächlich schon vor Ende des ersten Studienteils, und bevor Tests zu Nahrungsmittel-Nebenwirkungen und zur Pharmakokinetik und -dynamik begonnen hatten.
Am 6. Oktober 2015 begannen die ersten acht Probanden auf zehn aufeinanderfolgenden Tagen entweder 2,5 mg Wirkstoff zu nehmen, oder – je zwei Teilnehmer – ein Placebo. In vier weiteren Gruppen wurde die Dosis dann jeweils verdoppelt: Eine schwere Nebenwirkung wurde der ANSM während dieser vier ersten Gruppen des MAD-Teils nicht zur Kenntnis gebracht, so die Behörde.
Ab der fünften Gruppe gibt das Dokument die Werktage an: Am Mittwoch, den 6. Januar, erhielten die Teilnehmer der fünften MAD-Gruppe die erste Dosis von nun 50 mg. Am Sonntag, den 10. Januar, erhielten die acht Probanden zum fünften Mal diese Dosis, berichtet ANSM.
Später am selben Tag wurden dann bei einem Probanden „Symptome“ festgestellt, die abends zu einer Einweisung ins Krankenhaus führten, protokolliert ANSM nüchtern in ihrem Protokoll. Laut dem Leiter der Neurologie am Uniklinikum Rennes, Gilles Edan, waren die Symptome so stark, dass das Team zuerst von einem Schlaganfall ausging.
Abbruchkriterien zu liberal
Trotzdem erhielten am Montag (11. Januar) laut ANSM-Bericht die restlichen sieben Probanden der fünften MAD-Gruppe ihre sechste Dosis (genauer: fünf erhielten den Wirkstoff, zwei ein Placebo). Es sollte die letzte sein.
An diesem Tag wurde bei dem zuvor eingewiesenen Patienten, dessen Symptome sich verschlechtert hatten, ein Kernspin durchgeführt – und festgestellt, dass er ins Koma gefallen war. Der Mann war hier möglicherweise schon hirntot.
Nach einer ersten Einschätzung von Oliver Cornely, akademischer Leiter des Zentrums für Klinische Studien an der medizinischen Fakultät der Universität Köln, handelten die Verantwortlichen hier wohl im Einklang mit dem Studienprotokoll. Dieses sah vor, dass die Gabe der nächsten Dosis nur gestoppt werden solle, wenn bei vier oder mehr Probanden gleichartige schwerere Nebenwirkungen auftreten.
Da die Abbruchkriterien zu liberal waren, entsprach dieses Vorgehen dem Studienprotokoll, so Cornely. Erst später an diesem Montag wurde die klinische Studie „mit Zustimmung vom Sponsor Bial und dem Institut Biotrial“ „unterbrochen“, schreibt das ANSM.
Erst zwei bis vier Tage nach der Diagnose „Koma“ des ersten Betroffenen wurden die fünf weiteren Probanden, die den Wirkstoff noch erhalten hatten, in die Klinik eingewiesen. Und erst drei Tage nach Beendigung der Studie informierte Biotrial die Behörde über das Auftreten der schweren Nebenwirkungen, so ANSM.
Keine Lehre aus dem Tegenero-Fiasko?
Laut dem ersten Bericht der französischen Behörde hätte die Studie schon mit einem Verstoß gegen den Studienplan begonnen. In der ersten Phase erhielten wie beim MAD-Teil jeweils sechs von acht Probanden in insgesamt acht Gruppen den Wirkstoff, und zwei ein Placebo („Single Ascending Dose“, SAD).
Doch anders als im Protokoll vorgeschrieben, wurde laut Bericht am Mittwoch am 9. Juli 2015 zwei ersten Probanden der ersten Gruppe 0,25 mg BIA 10-2474 gegeben, vier weiteren am nächsten Tag. Das Studienprotokoll sah hingegen vor, am ersten Tag nur einem Probanden den Wirkstoff zu verabreichen, die restlichen sollten ihn 24 Stunden später bekommen. Auf Nachfrage von DAZ.online sagte ANSM am Freitag (29.1.), dass sie den Bericht in diesem Punkt korrigieren würde: Am ersten Tag hätte doch nur ein Proband den Wirkstoff bekommen, fünf weitere am nächsten.
Doch laut EMA-Guideline sollen in den ersten Kohorten, die eine neue Substanz erhalten, alle Probanden den Wirkstoff mit ausreichendem zeitlichem Abstanz zueinander bekommen, um ein Fiasko wie bei der parallelen Gabe von TGN1424 vor zehn Jahren in London zu verhindern. Nur in begründeten Ausnahmefällen solle hiervon abgewichen werden.
Die Arzneimittelbehörde ANSM hält in ihrem Bericht fest, dass sie im Oktober 2014 das Prüfinstitut Biotrial auf die Regeln der Guten Klinischen Praxis und im Dezember 2014 auf die Einhaltung der Guten Laborpraxis geprüft habe. Außerdem habe die regionale Gesundheitsbehörde ARS das Testzentrum mehrfach untersucht.
Den Antrag zur Genehmigung der Studie hätte Biotrial im Auftrag von Bial am 30. April 2015 eingereicht, dieser sei auf prä-klinische, klinische und pharmazeutische Qualität geprüft worden. Nachdem geforderte Ergänzungen nach mehrfachem Austausch nachgebessert wurden, habe die ANSM zwei Monate später zugestimmt, wie auch einige Tage später die zuständige Ethikkommission.
Durch das von ANSM veröffentlichte Protokoll wurde nun deutlich, dass die Studie trotz des Zwischenfalls am Tag darauf erst einmal fortgesetzt wurde. Dies könnte den lockeren Abbruchkriterien geschuldet sein, die von Experten scharf kritisiert wurden. Auch wurden die weiteren Probanden erst zwei bis vier Tage nach der Diagnose des Komas in die Klinik eingewiesen.
Für Ende März hat die Arzneimittelbehörde einen Abschlussbericht versprochen.
Update: Am Freitag, den 29.01.2016 erklärte die französische Arzneimittelbehörde ANSM auf Nachfrage von DAZ.online, dass ihr Bericht von Mittwoch in Bezug auf die Gabe des Wirkstoffs an die ersten sechs Patienten falsch war und korrigiert werde. Dieser Artikel wurde entsprechend ergänzt.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.