















- DAZ.online
- DAZ / AZ
- DAZ 52/2018
- Informationen über unerw...
Arzneimittelsicherheit
Informationen über unerwünschte Wirkungen
Rote-Hand-Brief zu Midazolam
Für Midazolam (Buccolam®) wurden mit einem erneuten Rote-Hand-Brief die risikominimierenden Maßnahmen verschärft, nachdem bei der Lösung zur Anwendung in der Mundhöhle zwei Berichte zu jeweils einer verschluckten beziehungsweise aspirierten durchscheinenden Verschlusskappe der Applikationsspritzen der Buccolam® Lösung gemeldet wurden. Der durchscheinend-weiße Verschluss sollte eigentlich zusammen mit der roten Schutzkappe entfernt werden. Verbleibt er jedoch versehentlich auf der Spritze, ist die Verabreichung der Lösung nicht möglich. Der innere durchscheinend-weiße Verschluss muss dann manuell entfernt werden, damit er nicht bei der Applikation in den Mund des Patienten gelangt. Apotheken sollten Patienten, Eltern und Betreuer informieren, an die das Antiepileptikum in der Vergangenheit abgegeben wurde. Allen zukünftigen Auslieferungen der Lösung wird der Sicherheitshinweis beiliegen. (DAZ 4, S. 119)
Stufenplanverfahren zu Dimenhydrinat- und Diphenhydramin-haltigen Antiemetika
Orale und rektale Dimenhydrinat- und Diphenhydramin-haltige Antiemetika sollen bei Kindern unter drei Jahren nur nach strenger Indikationsstellung und sorgfältiger Beachtung der Dosierung angewendet werden. Es besteht der begründete Verdacht, dass es insbesondere bei der Behandlung einer banalen Gastroenteritis oder eines fieberhaften Infektes sowie bei Überdosierung zu schweren Nebenwirkungen wie Krampfanfällen kommen kann. Die Texte der Fach- und Gebrauchsinformationen werden angepasst. Insbesondere wird auf die Gefahr von lebensbedrohlichen Überdosierungen (Festlegung einer Obergrenze für die Tagesdosis per kg Körpergewicht) und auf eine strenge Indikationsstellung hingewiesen. (DAZ 2, S. 104)
Foto: Tomsickova – stock.adobe.com
Rote-Hand-Brief zu Saccharomyces boulardii
Bei Saccharomyces-boulardii-haltigen Arzneimitteln wurden bei schwerkranken oder immunsupprimierten Patienten die Kontraindikationen erweitert. Arzneimittel mit Saccharomyces boulardii/Saccharomyces cerevisiae HANSEN-CBS-5926 sind unter anderem indiziert zur unterstützenden symptomatischen Behandlung von Diarrhöen, zusammen mit Rehydratation oder diätetischen Maßnahmen. Das Risiko der Fungämien bei Patienten mit zentralem Venenkatheter war bereits bekannt. Nachdem diese nun auch bei hospitalisierten, schwerkranken oder immunsupprimierten Patienten berichtet wurden, wurde nach Auswertung von 61 Fungämie-Fällen die Erweiterung der Anwendungsbeschränkungen empfohlen. Die Produktinformationen werden entsprechend angepasst. Da das Risiko der Kontamination mit S. boulardii groß ist, gilt für medizinisches Fachpersonal besondere Vorsicht bei der Handhabung in Gegenwart von schwerkranken oder immunsupprimierten Patienten sowie Patienten mit zentralem oder auch peripherem Venenkatheter. (DAZ 4, S. 120)
Patienten-Erinnerungskarte für Pamidronsäure
Aufgrund des Risikos einer Kiefernekrose während der Behandlung mit Bisphosphonat-haltigen Arzneimitteln empfahl der PRAC Anfang 2018, dass zusätzliche risikominimierende Maßnahmen erforderlich sind. Dazu gehört Schulungsmaterial wie eine Patienten-Erinnerungskarte, die insbesondere Patienten vor und während der Therapie mit Pamidronsäure-Infusionen auf das Risiko einer Osteonekrose aufmerksam macht. Die Patientenkarte soll Bestandteil jeder Verpackung Pamidronsäure-haltiger Arzneimittel sein, Ärzte und Apotheker sollen jedem Patienten bei Verordnung, Abgabe bzw. Verabreichung von Pamidronsäure-haltigen Durchstechflaschen die Patientenkarte aushändigen und erläutern. Der Patient wird hierin angehalten, eine gute Mundhygiene aufrechtzuerhalten, routinemäßige zahnärztliche Kontrolluntersuchungen durchführen zu lassen und sofort jegliche Symptome im Mund, wie z. B. Lockerung der Zähne, Schmerzen oder Schwellungen oder nicht verheilende Wunden oder Sekretaustritt im Mund oder Kieferbereich, zu berichten. (DAZ 4, S. 122)
Foto: kurousagi – stock.adobe.com
Rote-Hand-Brief zu Valproat-haltigen Arzneimitteln
Bei Kindern, die im Mutterleib Valproat ausgesetzt waren, besteht ein hohes Risiko für schwerwiegende Entwicklungsstörungen und angeborene Missbildungen. Schon 2014 wurden Warnhinweise und Anwendungsbeschränkungen dazu veröffentlicht. Nachdem zusätzlich Schulungsmaterialien angeordnet wurden, folgte 2017 die Einführung einer Patientenkarte als Bestandteil jeder Packung Valproat-haltiger Arzneimittel. Da diese Maßnahmen nicht effektiv waren, wurden nun weitere Maßnahmen eingeführt: Bei bipolaren Störungen und als Off-Label-Use in der Migräneprophylaxe sind Valproat-haltige Arzneimittel während der Schwangerschaft grundsätzlich kontraindiziert. Bei Epilepsie dürfen sie in der Schwangerschaft nur angewendet werden, wenn keine alternativen Behandlungen zur Verfügung stehen. Wenden trotz bestehender Kontraindikation Frauen im gebärfähigen Alter Valproat-haltige Arzneimittel an, muss eine zuverlässige Schwangerschaftsverhütung und eine Beratung durch den Arzt durchgeführt werden. Frauen im gebärfähigen Alter, die Valproat anwenden, sollten ihren Arzt aufsuchen, damit die Behandlung gegebenenfalls erneut beurteilt werden kann und um zu entscheiden, ob die Bedingungen des Schwangerschaftsverhütungsprogramms aktuell eingehalten werden. Apotheker sollen zudem bei Abgabe Valproat-haltiger Arzneimittel die Patientenkarte aushändigen. Die Produktinformationen werden entsprechend der Maßnahmen angepasst, auch die äußere Verpackung wird mit einem zusätzlichen umrandeten Warnhinweis versehen. (DAZ 46, S. 111)
Ruhen der Zulassung von Hydroxyethylstärke-haltigen Arzneimitteln
Der PRAC hat im Rahmen eines Risikobewertungsverfahrens das EU-weite Ruhen der Zulassung für Hydroxyethylstärke(HES)-haltige Arzneimittel zur Infusion empfohlen, nachdem Anwendungsstudien zeigten, dass Hydroxyethylstärke-haltige Arzneimittel immer noch außerhalb der Zulassung verwendet werden, wie bei kritisch kranken Patienten oder solchen mit Sepsis und Nierenschäden. Das Verfahren war bereist im Oktober 2017 eröffnet. Anwendungsbeschränkungen für entsprechende Patientengruppen wurden aufgrund von Nierenschäden und Todesfällen bereits im Jahr 2013 eingeführt. Im Hinblick auf die schwerwiegenden Risiken, denen bestimmte Patientengruppen ausgesetzt sind, hat der PRAC das Ruhen der Zulassungen HES-haltiger Arzneimittel empfohlen, zumal alternative Behandlungsoptionen verfügbar sind. (DAZ 4, S. 121)
Leberwertkontrolle bei Ulipristalacetat bei Uterusmyomen empfohlen
Mit einem Rote-Hand-Brief wurde über das Ende des Risikobewertungsverfahrens zu Ulipristalacetat 5 mg Tabletten (Esmya®) und zu Indikationseinschränkung, neuer Kontraindikation sowie der Überwachung der Leberfunktion informiert. Aufgrund des Risikos für schwere Leberschäden darf bei Frauen mit bestehender Leberfunktionsstörung Esmya® nicht angewendet werden. Eine Intervalltherapie mittlerer bis starker Symptome durch Gebärmutter-Myome darf nur bei Patientinnen erfolgen, für die eine Operation nicht infrage kommt. Esmya® bleibt weiterhin indiziert für ein einmaliges Behandlungsintervall von bis zu drei Monaten zur präoperativen Behandlung mittlerer bis starker Symptome durch Gebärmutter-Myome. Regelmäßig müssen Leberfunktionstests durchgeführt werden. Eine Patientenkarte zu den Hintergründen und der Planung von Leberfunktionstests ist in jeder Arzneimittelpackung enthalten. Bei Anzeichen und Symptomen einer Leberschädigung (Müdigkeit, Gelbfärbung der Haut, dunkler Urin, Übelkeit, Erbrechen) sollte unverzüglich der behandelnde Arzt konsultiert werden. (DAZ 7, S. 101; DAZ 8, S. 109 und DAZ 32, S. 102)
Foto: Gedeon Richter Pharma GmbH
Bei Mycophenolat keine zusätzliche Verhütung zwingend empfohlen
Mycophenolat dient der Verhinderung einer Transplantatabstoßung und ist stark genotoxisch. Sexuell aktiven Männern wurde bislang empfohlen, während der Behandlung und für mindestens 90 Tage nach Beendigung der Behandlung Kondome zu benutzen. Überdies sollten die Partnerinnen eine weitere Verhütungsmethode anwenden. Im Dezember 2017 wurde festgestellt, dass Daten bei Schwangerschaften, bei denen der Vater Mycophenolatmofetil(MMF)-/Mycophenolsäure-haltigen Arzneimittel (MPA) angewendet hat, nicht auf ein erhöhtes Risiko für Fehlgeburten und kongenitale Missbildungen hindeuten. Daher sei es nach Ansicht der EMA angemessen, eine zuverlässige Verhütung – entweder durch den männlichen Patienten oder durch dessen Partnerin – während und für mindestens 90 Tage nach Behandlungsende anzuwenden. Die Produktinformation und das Schulungsmaterial betroffener Arzneimittel werden entsprechend angepasst. Für Frauen im gebärfähigen Alter, die keine wirksame Verhütungsmethode anwenden, sind MMF beziehungsweise MPA weiterhin kontraindiziert. (DAZ 6, S. 111)
Obeticholsäure-Dosis an die Leberfunktion anpassen
Die Intercept Pharma GmbH informierte über Dosierungsempfehlungen von Obeticholsäure (Ocaliva®) bei Patienten mit mittelschwerer bis schwerer Einschränkung der Leberfunktion. Der Farnesoid-X-Rezeptor(FXR)-Agonist ist indiziert bei Patienten mit primär biliärer Cholangitis. Vor Behandlungsbeginn muss der Leberstatus bekannt sein, die Dosis ist bei Einschränkungen der Leberfunktion entsprechend anzupassen. (DAZ 7, S. 100)
Hypersensitivitätsreaktionen unter Flurbiprofen
Die AMK erinnerte an das Risiko für Hypersensitivitätsreaktionen bei Patienten, die Halsschmerzen mit Flurbiprofen-haltigen Rachentherapeutika behandeln. Das Arylpropionsäure-Derivat Flurbiprofen ist ein nichtsteroidales Antirheumatikum und wird seit 2004 vor allem im Rahmen der Selbstmedikation zur Therapie von Entzündungen und Schmerzen der Rachenschleimhaut bei Infektionen der oberen Atemwege ab einem Alter von zwölf Jahren eingesetzt. Derzeit werden in dieser Indikation Lutschtabletten und Rachensprays vermarktet, für die eine maximale Anwendungsdauer von drei Tagen empfohlen wird. Eine Analyse von an die AMK gemeldeten Verdachtsfällen zu Flurbiprofen-haltigen Arzneimitteln über einen Zeitraum von fünf Jahren ergab insgesamt 78 Berichte, von denen in 53 Fällen unerwünschte Arzneimittelwirkungen beobachtet wurden, die bei 16 Patienten schwerwiegend verliefen. Bei der Abgabe von Flurbiprofen-haltigen Arzneimitteln sollten sorgfältig Nutzen und Risiken gegeneinander abgewogen und Patienten über das Risiko von Hypersensitivitätsreaktionen beraten werden. Patienten mit einer bekannten NSAR-Unverträglichkeit sollten zur Behandlung von Halsschmerzen kein Flurbiprofen erhalten. Überdies ist Vorsicht geboten bei älteren Patienten sowie bei Patienten mit allergischem Asthma oder Allergien in der Anamnese. (DAZ 19, S. 112)
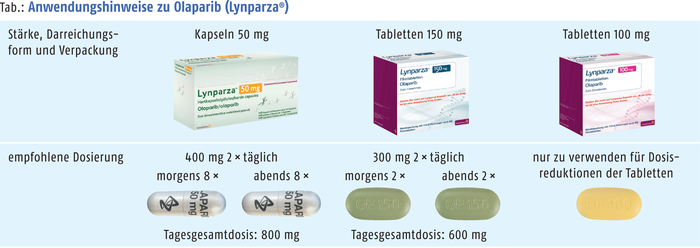
Rote-Hand-Brief zu Olaparib
Durch die Einführung einer neuen Darreichungsform bei Olaparib (Lynparza®) hat sich das Risiko von Medikationsfehlern erhöht. Lynparza® Kapseln sind bereits seit Dezember 2014 zentral zugelassen, Tabletten wurden im Mai 2018 eingeführt. Für die Darreichungsformen Tablette und Kapsel in den jeweiligen Anwendungsgebieten ergeben sich unterschiedliche Dosierschemata. Ein Austausch der Darreichungsformen ist aufgrund unterschiedlicher Bioverfügbarkeiten mit dem Risiko einer Über- beziehungsweise Unterdosierung verbunden, wenn dieser ausschließlich anhand der Dosis (Milligramm per Milligramm) erfolgt (siehe Abb.). Der PARP-Inhibitor als Tablette (100 mg oder 150 mg) ist indiziert zur Erhaltungstherapie Erwachsener mit Platin-sensitivem Rezidiv eines high-grade epithelialen Ovarial-, Eileiter- oder primären Peritonealkarzinoms. Lynparza® Kapseln (50 mg) sind zur Erhaltungstherapie Erwachsener mit Platin-sensitivem Rezidiv eines BRCA-mutierten high-grade serösen epithelialen Ovarial-, Eileiter- oder primären Peritonealkarzinoms indiziert. Apotheker werden gebeten, sicherzustellen, dass die ärztliche Verschreibung genaue Angaben zur Darreichungsform und Dosierung enthält und die korrekte Packung abgegeben wird. (DAZ 22, S. 96)
Widerruf der Zulassung Flupirtin-haltiger Arzneimittel
Die Zulassung aller Flupirtin-haltigen Arzneimittel wurde widerrufen, nachdem die Bewertung vorhandener Studienergebnisse und verfügbarer Daten zu Nutzen und Risiken aus klinischen Prüfungen sowie Fallberichten ergab, dass trotz der bislang eingeführten risikominimierenden Maßnahmen aus dem Jahr 2013, weiterhin Fälle schwerwiegender Leberschäden, einschließlich Leberversagen, auftraten. (DAZ 8, S. 110 und DAZ 23, S. 107)
Rote-Hand-Brief zu Fluorchinolon-haltigen Arzneimitteln
In einem Rote-Hand-Brief informierten mehrere Zulassungsinhaber über das erhöhte Risiko von Aortenaneurysmen und -dissektionen bei der systemischen und inhalativen Anwendung von Fluorchinolon-haltigen Arzneimitteln. In Deutschland sind Ciprofloxacin, Levofloxacin, Moxifloxacin, Norfloxacin und Ofloxacin zugelassen. Bei Patienten mit einem Risiko für Aortenaneurysmen und -dissektionen sollten Fluorchinolone nur nach sorgfältiger Nutzen-Risiko-Abwägung angewendet werden. Insbesondere bei plötzlich auftretenden schweren Bauch-, Brustkorb- oder Rückenschmerzen sollten Patienten unverzüglich in der Notaufnahme ärztliche Hilfe in Anspruch nehmen. (DAZ 44, S. 113)
Hautkrebs-Risiko unter Hydrochlorothiazid
Die Zulassungsinhaber von Hydrochlorothiazid(HCT)-haltigen Arzneimitteln informierten in einem Rote-Hand-Brief über das Risiko von nichtmelanozytärem Hautkrebs (non-melanoma skin cancer, NMSC). Zwei neue pharmakoepidemiologische Studien aus Dänemark zeigten ein erhöhtes Risiko für Basalzellkarzinom (Basaliom) und Plattenepithelkarzinom der Haut (Spinaliom) bei Exposition mit steigenden kumulativen Dosen von HCT. Danach könnte sich das Risiko in Abhängigkeit der kumulativen Dosis von HCT um das 4- bis 7,7-Fache für Spinaliome bzw. um das 1,3-Fache für Basaliome erhöhen. Als möglicher Mechanismus wird die photosensibilisierende Wirkung von HCT diskutiert. Patienten sollten über das Risiko informiert werden, ihre Haut regelmäßig auf Hautveränderungen untersuchen und auf einen angemessenen Sonnenschutz achten. (DAZ 43, S. 110)
Rückruf Valsartan-haltiger Arzneimittel
Die AMK informierte mehrfach über die Rückrufe verschiedener Valsartan-haltiger Arzneimittel mit verunreinigtem Wirkstoff (u. a. DAZ 28, S. 113 und 114, DAZ 29, S. 115). Anlässlich der Empfehlung des BfArM, Patienten unter ärztlicher Kontrolle auf nicht betroffene Arzneimittel umzustellen, publizierte die AMK eine Vergleichstabelle, die bei der Abschätzung einer entsprechenden Äquivalenzdosis eines alternativen Sartans zur bisherigen Valsartan-Dosis helfen kann. Da Lieferengpässe für Valsartan und andere Sartane nicht ausgeschlossen werden können, wurde die ursprüngliche Vergleichstabelle auf ACE-Hemmer ausgeweitet. Ausdrücklich weist die AMK darauf hin, dass diese Tabellen nur einen Anhaltspunkt darstellen und wirkstoffbezogene bzw. patientenindividuelle Faktoren beachtet werden sollten. (DAZ Nr. 29, S. 116; DAZ 30, S. 99)
Psychische Symptome und sexuelle Dysfunktion unter Finasterid
Der 5-α-Reduktase-Inhibitor Finasterid ist bei androgenetischer Alopezie (Tagesgesamtdosis 1 mg) sowie bei benigner Prostatahyperplasie (Tagesgesamtdosis 5 mg) indiziert. Unter Finasterid wurde über Stimmungsänderungen, depressive Verstimmung, Depression und Suizidgedanken, sexuelle Dysfunktion, einschließlich erektiler Dysfunktion, Ejakulationsstörung und verminderte Libido berichtet. Sexuelle Dysfunktionen können nach dem Absetzen länger als zehn Jahre fortbestehen. Die Fach- und Gebrauchsinformationen wurden entsprechend aktualisiert und die Nebenwirkung „Angst“ sowie ein neuer Warnhinweis für alle Finasterid-haltigen Arzneimittel ergänzt. Zudem wurde für die 1-mg-Stärke die Nebenwirkung „depressive Verstimmung“ in „Depression“ geändert. (DAZ 27, S. 109)
Foto: RFBSIP – stock.adobe.com
Fastjekt® – Haltbarkeit einiger Chargen verlängert
Die Firma Meda Pharma GmbH & Co. KG informierte über die Verlängerung des Verfalldatums bestimmter Chargen von Fastjekt® 300 µg, Injektionslösung in einem Fertigpen, um vier Monate. Patienten können die entsprechenden Autoinjektoren bis zum verlängerten Verfalldatum nutzen. So soll die Verfügbarkeit des seit Juli 2018 von einem Lieferengpass betroffenen Arzneimittels zur Notfallbehandlung einer schweren allergischen Reaktion kurzfristig verbessert werden. Das BfArM hatte Unterlagen zur pharmazeutischen Qualität geprüft und kam zu dem Schluss, dass durch die Verlängerung des Verfalldatums kein erhöhtes Gesundheitsrisiko für Patienten entsteht. Die Empfehlung zur Verlängerung der Verfalldaten gilt nicht für den Fastjekt® Junior 150 µg, Injektionslösung in einem Fertigpen. (DAZ 36, S. 101)
Minimierung des teratogenen Risikos von Sonidegib
Der Hedgehog-Signalweg-Inhibitor Sonidegib (Odomzo®) wird zur Behandlung Erwachsener mit lokal fortgeschrittenem Basalzellkarzinom (BCC) eingesetzt, die für eine kurative Operation oder eine Strahlentherapie nicht infrage kommen. Da Sonidegib embryotoxisch und/oder teratogen ist und bei schwangeren Frauen zum embryofetalen Tod oder schweren Geburtsfehlern führen kann, darf das Arzneimittel während der Schwangerschaft nicht angewendet werden. Bei Frauen im gebärfähigen Alter sind innerhalb von sieben Tagen vor sowie während der Behandlung monatlich Schwangerschaftstests durchzuführen. Frauen müssen während der Therapie und noch 20 Monate nach der letzten Dosis zwei empfohlene Methoden der Schwangerschaftsverhütung anwenden. Männliche Patienten müssen während der Einnahme des antineoplastischen Mittels und noch sechs Monate nach Behandlungsende beim Geschlechtsverkehr ein Kondom benutzen. Apotheker sollten die Patienten informieren und auf entsprechendes Schulungsmaterial (Patienteninformationsbroschüre mit der Patienten-Erinnerungskarte und dem Beratungsnachweisformular) hinweisen. (DAZ 10, S. 118)
Neuralrohrdefekte bei Neugeborenen unter Dolutegravir
In einem Rote-Hand-Brief wurde über das erhöhte Risiko für Neuralrohrdefekte bei Neugeborenen informiert, deren Mütter zur Zeit der Konzeption Dolutegravir (Tivicay®), Dolutegravir/Abacavir/Lamivudin (Triumeq®) oder Dolutegravir/Rilpivirin (Juluca®) angewendet haben. Dolutegravir hemmt die Integrase des humanen Immundefizienzvirus (HIV). In einer Anwendungsbeobachtung wurden insgesamt vier Fälle von Neuralrohrdefekten aus einer Gruppe von 426 Neugeborenen registriert, wenn Frauen Dolutegravir einnahmen. Bei Frauen im gebärfähigen Alter sollte vor Beginn einer Therapie mit Dolutegravir eine bestehende Schwangerschaft ausgeschlossen werden. Unter einer Therapie sollten Frauen während der gesamten Behandlung wirksam verhüten. Frauen mit Kinderwunsch sollten kein Dolutegravir erhalten. Die Fachinformationen der betroffenen Präparate werden aktualisiert. (DAZ 23, S. 106)
Rote-Hand-Brief zu Cefepim
Die Firma Bristol-Myers Squibb informierte im einem Rote-Hand-Brief über das Risiko schwerer Nebenwirkungen unter Cefepim (Maxipime®) bei Patienten mit reduzierter Nierenfunktion, wenn höhere Dosen als empfohlen gegeben werden. Das parenteral zu verabreichende Betalaktam-Antibiotikum aus der Gruppe der Cephalosporine wird fast ausschließlich über renale Wege ausgeschieden. Bei Patienten mit eingeschränkter Nierenfunktion (Kreatinin-Clearance ≤ 50 ml/min), die das Cephalosporin in einer höheren als der empfohlenen Dosis erhielten, kam es zu Fällen von Bewusstseinsstörungen mit Verwirrtheit, Halluzinationen, Stupor und Koma, Myoklonus oder Krampfanfällen. Neben älteren Patienten waren auch Patienten mit normaler Nierenfunktion bei Überschreitung der empfohlenen Dosierung betroffen. Der Hersteller erinnerte an eine Dosisanpassung gemäß Fachinformation. Auch muss bei Kombination mit potenziell nephrotoxischen Antibiotika, wie Aminoglykosiden oder starken Diuretika, die Nierenfunktion sorgfältig überwacht werden. (DAZ 25, S. 107)
Über- oder Unterdosierung Methotrexat-haltiger Fertigspritzen
Das BfArM hat 16 Fallberichte zu Methotrexat(MTX)-haltigen Fertigspritzen ausgewertet, die im Zusammenhang mit einer möglichen unabsichtlichen Über- oder Unterdosierung standen. Einmal wöchentlich, niedrig dosiert, wirkt Methotrexat immunsuppressiv und zählt zu den antirheumatischen Basistherapeutika, vor allem in der Dermatologie (Psoriasis vulgaris und Psoriasis arthropathica) und Rheumatologie (rheumatoide Arthritis). Als Hauptursache für Fehldosierungen konnten Beinahe- und/oder potenzielle Medikationsfehler identifiziert werden, die durch die Art und Weise der Deklaration mitbedingt wurden. So können beispielsweise identisch bezeichnete Arzneimittel unterschiedliche Mengen an Wirkstoff in verschiedenen Füllvolumina in einer Fertigspritze enthalten. Grundsätzlich sind MTX-haltige Arzneimittel unterschiedlicher Zulassungsinhaber, deren absoluter MTX-Gehalt pro Fertigspritze gleich ist, über Rabattverträge austauschbar, doch die Deklaration der auszutauschenden Arzneimittel kann sich unterscheiden. Unterschiedliche Angaben wie zum Beispiel Abfolgen von Konzentration und Dosis auf dem Umkarton und in der Praxis/Apotheken-Software, können Verordnungsfehler und Falschabgaben begünstigen oder Patienten verwirren. Die AMK bittet Apotheker, MTX-Verordnungen kritisch zu prüfen, achtsam bei der Auswahl und Abgabe zu sein und Patienten zu möglichen Abweichungen bei der Deklaration ausgetauschter Packungen zu beraten. (DAZ 27, S. 110)
Foto: Bayer
Hinweis auf das lebertoxische Risiko unter Iberogast®
Das BfArM informierte, dass die Bayer Vital GmbH für das Schöllkraut-haltige Arzneimittel Iberogast® die im Stufenplanbescheid angeordneten Änderungen der Produktinformationen in Bezug auf das lebertoxische Risiko umsetzt. In Deutschland hatte der Zulassungsinhaber Klage eingereicht, sodass die Hinweise auf Lebertoxizität bislang nicht in die Fach- und Gebrauchsinformation eingefügt wurden. Das pflanzliche Kombinationspräparat enthält Extrakte aus neun Heilpflanzen, unter anderem auch Schöllkrautextrakt. Hintergrund sind neue bekannt gewordene Nebenwirkungsmeldungen von Leberschädigungen im Zusammenhang mit Iberogast®. Bei bestehenden Lebererkrankungen oder gleichzeitiger Anwendung von Arzneimitteln mit leberschädigenden Eigenschaften darf das Arzneimittel nicht eingenommen werden. Besondere Vorsicht bei der Einnahme von Iberogast® ist erforderlich, wenn Zeichen einer Leberschädigung (Gelbfärbung der Haut oder Augen, dunkler Urin, entfärbter Stuhl, Schmerzen im Oberbauch, Übelkeit, Appetitverlust, Müdigkeit) auftreten. Die Einnahme ist sofort zu beenden und ein Arzt aufzusuchen. (DAZ 38, S. 103)
Aortitis unter Filgrastim, Pegfilgrastim, Lipegfilgrastim und Lenograstim
In Zusammenhang mit einer Behandlung mit Granulozyten-koloniestimulierenden Faktoren (G-CSF) wurden Fälle einer Entzündung der Aorta (Aortitis) beschrieben. Betroffen waren sowohl Tumorpatienten als auch gesunde Spender von Blutstammzellen. Kennzeichnend sind u. a. unspezifische Symptome wie Fieber, abdominale Schmerzen, Rückenschmerzen sowie erhöhte Entzündungsmarker, die nach Absetzen der G-CSF-haltigen Therapie abklangen. Ein kausaler Zusammenhang zwischen Aortitis und Filgrastim-, Pegfilgrastim-, Lipegfilgrastim- und Lenograstim-haltigen Arzneimitteln wird als möglich erachtet. Auf das seltene, potenziell aber lebensbedrohliche Risiko wird in den aktualisierten Produktinformationen verwiesen. (DAZ 26, S. 101)
Kein Daclizumab bei multipler Sklerose
Der humanisierte monoklonale Anti-CD25-Antikörper Daclizumab (Zinbryta®) ist zugelassen zur Behandlung der schubförmigen multiplen Sklerose. Nach Meldungen über Fälle von immunvermittelter Enzephalitis und Meningoenzephalitis bei Patienten, die mit dem Antikörper behandelt werden, wurde ein Risikoverfahren eingeleitet. Als Konsequenz empfiehlt die EMA das sofortige Ruhen der Zulassung und den Rückruf aus dem EU-Markt: Es sollen keine neuen Patienten mit Zinbryta® behandelt werden. Patienten sollen darüber aufgeklärt werden, dass wegen der langen Halbwertszeit des Arzneimittels Nebenwirkungen (z. B. anhaltendes Fieber, schwere Kopfschmerzen, Müdigkeit, Gelbsucht, Übelkeit oder Erbrechen) auch bis zu sechs Monate nach dem Absetzen auftreten können. (DAZ 10, S. 119; DAZ 11, S. 142)
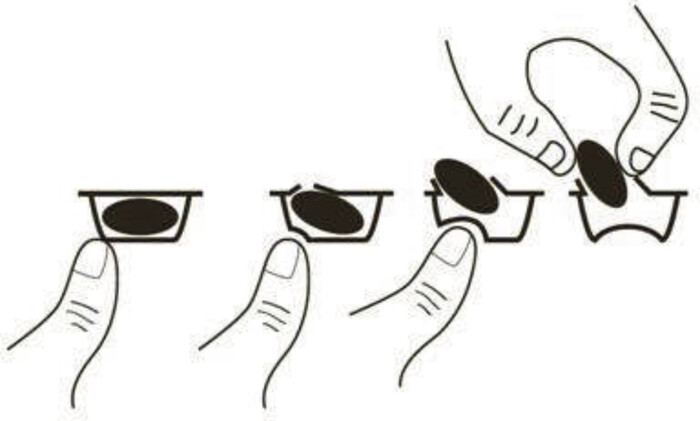
Minimierung des Kontaminationsrisikos bei Lenalidomid
Die Firma Celgene GmbH informierte über die sichere und korrekte Handhabung von Lenalidomid (Revlimid®, ▼). Der antineoplastische, antiangiogene, antiinflammatorische und immunmodulierende Wirkstoff wird zur Behandlung des multiplen Myeloms, myelodysplastischer Syndrome und des Mantelzell-Lymphoms eingesetzt. Da Lenalidomid mit dem teratogen wirkenden Thalidomid strukturverwandt ist, müssen strengste Sicherheitsmaßnahmen im Umgang mit Lenalidomid-haltigen Arzneimitteln beachtet werden. Der AMK wurden über einen Zeitraum von acht Jahren 22 Meldungen zu ausgetretenem Pulver bei Revlimid®-Kapseln gemeldet, wobei am häufigsten (13 Fälle) das Pulver beim Ausblistern freigesetzt wurde. Der Hersteller weist darauf hin, dass dies vermieden werden könne, wenn beim Ausblistern der Druck mit den Fingern auf das Kapselende, statt auf die Kapselmitte ausgeübt werden würde (siehe Abbildung). (DAZ 43, S. 110)
Foto: Celgene GmbH
Korrekte Handhabung von One-Point-Cut-Ampullen
Apotheken berichteten vereinzelt über Schwierigkeiten beim Öffnen/Brechen der One-Point-Cut(OPC)-Ampullen. Eine OPC-Ampulle hat am Ampullenhals eine Sollbruchstelle, die oberhalb mit einem Farbpunkt markiert wird. In einer Meldung an die AMK ließen sich die Ampullen nicht am markierten Punkt aufbrechen oder zerbrachen komplett. Zur korrekten Handhabung der OPC-Ampullen gibt es Hinweise sowie Piktogramme, die einem Informationsschreiben auf der AMK-Homepage zu entnehmen sind. (DAZ 38, S. 103)
Risiko venöser Thromboembolien bei Dienogest/Ethinylestradiol-haltigen Kontrazeptiva
Dass kombinierte hormonale Kontrazeptiva das Risiko für venöse Thromboembolien (VTE) im Vergleich zur Nichtanwendung erhöhen, ist bekannt. Jenapharm informierte im Dezember über neue Ergebnisse einer Metaanalyse von vier Beobachtungsstudien zum Risiko venöser Thromboembolien unter Dienogest/Ethinylestradiol-haltigen Kontrazeptiva. Daraus geht hervor, dass venöse Thromboembolien unter Dienogest nicht wie bislang vermutet ähnlich häufig wie unter Levonorgestrel-haltigen Präparaten auftreten. Das VTE-Risiko ist höher als gedacht und bewegt sich im Bereich anderer Gestagene der dritten und vierten Generation. Im Vergleich zu kombinierten hormonalen Kontrazeptiva, die Levonorgestrel enthalten, ergab die Metaanalyse für Dienogest-haltige Präparate ein 1,6-fach erhöhtes Risiko. Auch in den Fachinformationen der Dienogest-haltigen Präparate soll explizit auf das erhöhte Risiko hingewiesen werden. (DAZ 51, S. 105)
Liste „Bedenkliche Rezepturarzneimittel“ aktualisiert
Die AMK beurteilt regelmäßig Rezepturarzneimittel und aktualisiert die Liste bedenklicher Stoffe/Rezepturen. Im Mai 2018 wurde die Version vom Mai 2015 überarbeitet. Neu ist unter anderem, dass Kava-Kava/Piper methysticum aus der Liste gestrichen wurde. Der Zulassungswiderruf von 2007 wurde 2014 aufgehoben. Dem potenziellen Nutzen Kava-Kava-haltiger Arzneimittel stehen aber potenzielle Anwendungsrisiken gegenüber, weshalb die Zulassungen auf der Grundlage der Empfehlungen der Kommission E in mehreren Punkten angepasst wurden. Kava-Kava-Wurzelstock und seine Zubereitungen – ausgenommen in homöopathischen Zubereitungen zur oralen Anwendung, die nach der Herstellungsvorschrift 26 des Homöopathischen Arzneibuches hergestellt sind – unterliegen nun der Verschreibungspflicht nach AMVV. (DAZ 23, S. 108)
Vorkommnisse bei Medizinprodukten nur ans BfArM melden
Da bei der AMK immer noch Spontanberichte zu Vorkommnissen von Medizinprodukten aus Apotheken eintreffen (279 im Jahr 2017), weist sie darauf hin, dass seit Anfang 2017 die Apotheken verpflichtet sind, alle Meldungen zu Vorkommnissen bei Medizinprodukten ausschließlich an das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) zu melden. Der AMK fehlt seither die rechtliche Grundlage zur Bearbeitung entsprechender Berichtsbögen. Auf den Seiten des BfArM www.bfarm.de im Bereich Service stehen offizielle Formulare für die Meldung von Vorkommnissen bei Medizinprodukten zur Verfügung. (DAZ 15, S. 88)
Anstieg von Gesamtmortalität und Blutungen unter Rivaroxaban
Die Bayer AG informierte mittels Rote-Hand-Brief über den Anstieg von Gesamtmortalität sowie Thromboembolie- und Blutungsereignissen bei Patienten, die nach einem kathetergestützten, perkutanen Aortenklappenersatz (Transcatheter Aortic Valve Implantation; TAVI) mit Rivaroxaban (Xarelto®) behandelten wurden gegenüber der vergleichenden Therapie mit Clopidogrel. Dies ergaben vorläufige Analysen einer multizentrischen klinischen Phase-III-Studie (GALILEO), die aufgrund dieser Ergebnisse vorzeitig abgebrochen wurde. Rivaroxaban ist nicht zur Thromboseprophylaxe bei Patienten mit künstlichen Herzklappen, inklusive TAVI, zugelassen und sollte bei diesen Patienten auch nicht angewendet werden. (DAZ 41, S. 134)
Foto: detailblick-foto – stock.adobe.com
Starkes Nasenbluten unter Sinupret®
Bei der AMK gingen von März 2000 bis Ende November 2017 insgesamt 18 Berichte aus Apotheken zu überwiegend schwerem Nasenbluten unter der Einnahme von Sinupret® forte und Sinupret® extract ein. Die Zahl der Meldungen nahm nach der Markteinführung von Sinupret® extract 2012 zu. Die Arzneimittel enthalten ein Pulvergemisch aus Enzianwurzel, Primel- und Holunderblüten sowie Ampfer- und Eisenkraut (Sinupret® forte) bzw. einen getrockneten ethanolischen Auszug hieraus (Sinupret® extract). Nasenbluten ist bislang weder als Nebenwirkung in der Fach- und Gebrauchsinformation aufgeführt noch in klinischen Studien beobachtet worden. In den gemeldeten Fällen wurde das Nasenbluten zweimal mit „extrem“ bzw. „sehr stark“ und in acht weiteren Fällen mit „stark“ beschrieben. Für die Kausalität spricht der enge zeitliche Zusammenhang sowie die Tatsache, dass in der Hälfte der Fälle Sinupret® das einzig genannte Arzneimittel war. Überdies führte das Nasenbluten in 16 Fällen zu einem Ende der Therapie, woraufhin die Blutung in allen Fällen aufhörte. Dennoch könnte auch die Sinusitis selbst ursächlich an den Blutungen beteiligt sein. Die AMK empfiehlt Apothekern, gemeinsam mit dem Patienten den Nutzen und die potenziellen Risiken der Behandlung abzuwägen. Tritt Nasenbluten auf, sind die Phytopharmaka abzusetzen und ein Arzt zu konsultieren. (DAZ 45, S. 103)
Warnhinweise bei gleichzeitiger Gabe von Opioiden und Z-Substanzen
In den Produktinformationen Benzodiazepin-haltiger bzw. -ähnlicher (z. B. Z-Substanzen) Arzneimittel werden die Warnhinweise zur gleichzeitigen Anwendung mit Opioiden aktualisiert. Die kombinierte Anwendung erhöht aufgrund additiver ZNS-dämpfender Effekte das Risiko für Atemdepression, Koma und Tod. Es wird darauf hingewiesen, dass die gleichzeitige Anwendung von Opioiden und Benzodiazepinen nur angebracht ist, wenn es keine alternativen Behandlungsmöglichkeiten gibt. Ist eine gleichzeitige Verschreibung notwendig, ist die niedrigste wirksame Dosis zu verwenden, die Behandlungsdauer sollte so kurz wie möglich sein. (DAZ 16, S. 105)
Risiko eines primären Malignoms unter Denosumab
Die Amgen GmbH informierte über Ergebnisse einer gepoolten Analyse von vier klinischen Phase-III-Studien, wonach bei Behandlung von Patienten mit fortgeschrittenen Krebserkrankungen und Knochenbefall mit Denosumab (Xgeva®) häufiger neue primäre Malignome berichtet wurden, als unter Zoledronsäure. Der humane monoklonale anti-RANK-Ligand-Antikörper Denosumab ist zugelassen zur Prävention skelettbezogener Komplikationen bei Erwachsenen mit fortgeschrittenen Krebserkrankungen und Knochenbefall sowie zur Behandlung von Riesenzelltumoren des Knochens, die nicht resezierbar sind. Die Fachinformation wird entsprechend aktualisiert. (DAZ 21, S. 90)
Verblasste Messskala bei Xyrem®-Dosierspritzen
Nach mehrfacher Anwendung der Dosierspritzen von Xyrem® (Natriumoxybat) kann die Messskala unleserlich werden, so dass Über- oder Unterdosierungen möglich sind. Das Betäubungsmittel ist zur Behandlung von Narkolepsie mit Kataplexie bei Erwachsenen indiziert. In den letzten drei Jahren erhielt der Hersteller UCB Pharma insgesamt elf Beanstandungen, wonach die Skala auf der Dosierspritze nach mehrfachem Gebrauch unleserlich geworden oder ganz verblasst war. Apotheker sollten bei jeder Abgabe auf die Möglichkeit des Verblassens der Messskala auf den Dosierspritzen hinweisen. Eine fehlerhafte Dosierspritze sollte unmittelbar gegen eine neue getauscht werden, die kostenfrei bei der Firma angefordert werden kann. Voraussichtlich ab Juni 2019 wird die Firma eine neue Dosierspritze einführen. Die AMK empfiehlt Apotheken, die Patienten mit Xyrem® versorgen, eine Dosierspritze im Voraus vorrätig zu halten. (DAZ 51, S. 106)
Sachverständigenausschuss für Verschreibungspflicht
Am 3. Juli 2018 fand die 79. Sitzung des Sachverständigen-Ausschusses für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes (AMG) im BfArM statt. Der Sachverständigen-Ausschuss empfiehlt, Zubereitungen zur dermalen Anwendung aus Natriumbituminosulfonat und Hydrocortisonacetat aus der Verschreibungspflicht zu entlassen. Die gleiche Empfehlung wurde für orales Levocetirizin und Diclofenac zum äußeren Gebrauch als Pflaster ausgesprochen.
Distickstoffmonoxid soll der Verschreibungspflicht unterstellt werden. Der Antrag, Methocarbamol aus der Verschreibungspflicht zu entlassen, wurde einstimmig abgelehnt. Wenn der Verordnungsgeber den Voten des Ausschusses folgt, können die Änderungen zum 1. Januar 2019 in Kraft treten. (DAZ 28, S. 113)
Änderung in der Verschreibungs- und Apothekenpflicht
Am 28. September 2018 wurde die Verordnung zur Änderung der Arzneimittelverschreibungsverordnung (AMVV) und die Verordnung über apothekenpflichtige und freiverkäufliche Arzneimittel (AMVerkRV) im Bundesgesetzblatt veröffentlicht. Die wichtigsten Informationen daraus: Durch Änderung der Position Ibuprofen werden bestimmte Kombinationen zur oralen Anwendung von Ibuprofen (in maximaler Einzeldosis von 400 mg und in einer maximalen Tagesdosis von 1200 mg) und Coffein (in maximaler Einzeldosis von 100 mg und in einer maximalen Tagesdosis von 300 mg) zur Behandlung von akuten mäßig starken Schmerzen bei Erwachsenen aus der Verschreibungspflicht entlassen. „Doxylamin – zur Behandlung von Schlafstörungen bei Kindern bis zum Alter von 18 Jahren“ sowie 26 weitere Positionen werden der Verschreibungspflicht unterstellt.
Die Anlage 1a der AMVerkRV weist Stoffe und Zubereitungen aus Stoffen aus, die weder der Verschreibungs- noch der Apothekenpflicht unterliegen. Hier werden Fertigarzneimittel neu gelistet, die als traditionelle pflanzliche Arzneimittel registriert sind und folgende Wirkstoffe oder Zubereitungen enthalten: „Goldrutenkraut/Echtes Goldrutenkraut und Zubereitungen“, „Orthosiphonblätter und Zubereitungen“ sowie „Birkenblätter und Zubereitungen“, auch in Mischungen und jeweils auch mit Zusatz arzneilich nicht wirksamer Stoffe oder Zubereitungen.
In Anlage 4 der AMVerkRV wurde der die Apothekenpflicht begründende Grenzwert für Arsen in Heilwässern weiter gesenkt. In Flaschen abgefüllte Heilwässer, die nun je Liter 0,01 mg Arsen oder mehr enthalten, unterliegen der Apothekenpflicht. (DAZ 41, S. 134)



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.