













- DAZ.online
- DAZ / AZ
- DAZ 39/2004
- Pathogenese und Therapie ...
Neuropathologie
Pathogenese und Therapie des Schlaganfalls
Foto: DAZ Archiv
Ein Schlaganfall wird in ca. 85% der Fälle durch den Verschluss einer großen Arterie und zu 15% durch Blutungen im Gehirn ausgelöst und führt zur Unterbrechung der Durchblutung im Gehirn (Ischämie). Es kommt zu einem schwerwiegenden Mangel an Sauerstoff und Glucose im betroffenen Bereich, damit zu einem raschen Abfall der Energiereserven in den Neuronen und zum Zusammenbruch der physiologischen, lebenserhaltenden Abläufe in den Zellen.
Nekrose und Apoptose nach zerebraler Ischämie
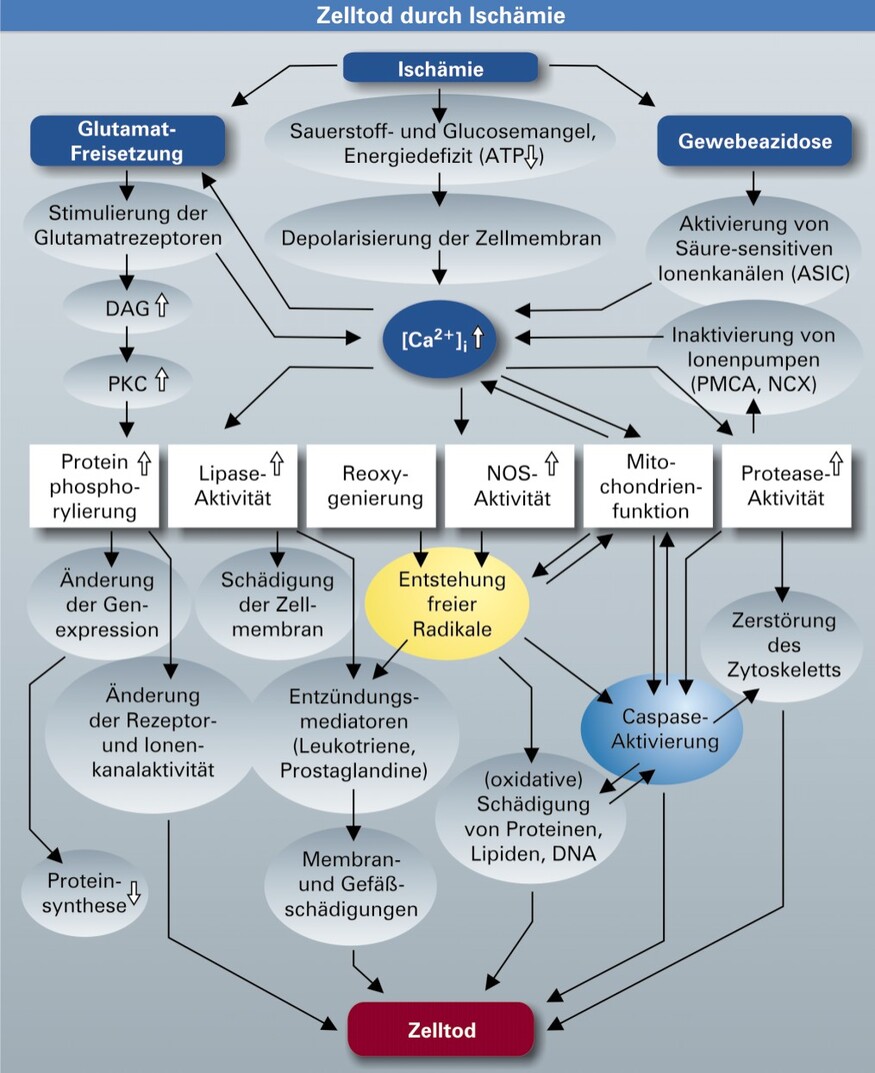
Bei zerebraler Ischämie werden verschiedene Mechanismen aktiviert, die unmittelbar oder verzögert zum unwiederbringlichen Verlust von Nervenzellen und damit zum Verlust von Gehirnfunktionen führen (Abb. 1). In einer frühen Phase kommt es zu einer massiven Freisetzung des erregenden Neurotransmitters Glutamat und zu fortgesetzten unkontrollierten elektrischen Entladungen (Depolarisationswellen). Darauf entgleist in den Nervenzellen die Ionenhomöostase, insbesondere steigt die Calciumkonzentration an.
Im weiteren Verlauf des Infarkts führen zwei Hauptmechanismen zum Absterben der Neuronen: Nekrose und Apoptose.
- induziert eine Entzündungsreaktion im Gehirngewebe, die zur schnellen Ausbreitung der Gewebeschädigung beiträgt.
- Die Apoptose ist im Gegensatz zur Nekrose ein aktiver, energieabhängiger Prozess, bei dem ein Programm zum "stillen" Selbstmord der Zellen aktiviert wird. Eine massive Entzündungsreaktion bleibt hier aus, und die Fragmente der sterbenden Zellen werden von benachbarten Zellen aufgenommen. Die Apoptose dominiert im Randbereich des Infarkts, in der Penumbra, wo sie über Stunden und Tage zu einer sekundären Ausbreitung der Gewebeschädigung führt.
- Nach neueren Erkenntnissen treten nach einem Schlaganfall auch Mischformen des Zelltods auf, bei denen die initiale Apoptose in eine sekundäre Nekrose übergeht.
Grafiken: Bühler
DAG = Diacylglycerol, PKC = Proteinkinase C, NOS = NO-Synthase.
Tödlicher Anstieg der Calciumkonzentration
Je nach Schweregrad der Schädigung und den Bedingungen in der Zelle können unterschiedliche Signalkaskaden aktiviert und entsprechende "Todesroutinen" induziert werden. Der exzessive Anstieg der intrazellulären Calciumkonzentration spielt dabei eine entscheidende Rolle. Dadurch kommt es in den Neuronen zur unkontrollierten Aktivierung von Protein- und DNA-spaltenden Enzymen sowie zur Schädigung von Mitochondrien, die zunächst Calcium vermehrt aufnehmen, dann aber ihre Fähigkeit zur Calciumspeicherung verlieren (P. Nicotera, Leicester).
Neben den bereits bekannten spannungsabhängigen und rezeptorgesteuerten Calciumkanälen sind erst kürzlich Azidose-sensitive Ionenkanäle (ASIC) identifiziert worden, die bei einem Abfall des extrazellulären pH-Wertes, wie er nach zerebraler Ischämie auftritt, durch Protonen (H+) aktiviert werden und Calcium in die Zelle strömen lassen (R. Simon, Portland). Weitere Untersuchungen müssen nun zeigen, ob die Blockade der ASIC nach einem Schlaganfall die Gewebeschädigung reduziert.
Calcium spielt auch eine Rolle beim Übergang von apoptotischen Mechanismen zur sekundären Nekrose. Offenbar werden in apoptotischen Zellen membranständige Calcium-ATPasen (PMCA), die als auswärts gerichtete Calciumionenpumpen fungieren, enzymatisch gespalten. Daneben ist die Spaltung des Natrium-Calcium-Austauschers (NCX) für den massiven Anstieg der intrazellulären Calciumkonzentration verantwortlich, der schließlich in die sekundäre Nekrose mündet. Die Spaltung und Inaktivierung des NCX erfolgt unter anderem durch Caspasen.
Caspase-Inhibitoren nur begrenzt wirksam
Caspasen sind Proteasen, die im Verlauf der Apoptose aktiviert werden und in einer hochgeordneten Kaskade weitere proapoptotische Faktoren in den Neuronen aktivieren. Die Hemmung von Caspasen ist daher eine wirkungsvolle Strategie, um die ischämische Schädigung von Hirngewebe deutlich zu reduzieren, doch reicht sie allein nicht aus, um den ischämischen Zelltod vollständig zu verhindern.
In transgenen Mäusen, die keine Caspase-3 exprimieren, ist zwar eine Verringerung des Infarktvolumens zu beobachten, dennoch stirbt ein Großteil der Neuronen im Randbereich des Infarktes weiterhin verzögert und mit typischen Anzeichen einer Apoptose ab. Zudem können Caspase-Inhibitoren in geschädigten Neuronen offenbar nicht die Degeneration von Dendriten und Axonen aufhalten, die unter anderem für die Verschaltung der Neuronen untereinander und damit für die komplexen Leistungen des Gehirns entscheidend sind.
PP2C-Hemmer gegen Atherosklerose und Infarkt?
Weitere Schlüsselfaktoren für die Degeneration des Gehirngewebes in einer frühen ischämischen Phase sind bestimmte Proteine der Bcl-2-Familie, die nach ihrer Aktivierung die Funktion der Mitochondrien direkt oder indirekt stören (Abb. 2). Zu diesen proapoptotischen Faktoren gehören unter anderem Bid, Bax und Bad, während andere Mitglieder der Bcl-2-Familie wie Bcl-2 oder Bcl-xL antiapoptotisch wirken. Bad wird durch Dephosphorylierung aktiviert. Dadurch löst es sich aus dem Komplex mit dem zytosolischen 14-3-3-Protein und bindet nunmehr an das antiapoptotische Bcl-xL.
Eine vermehrte Phosphorylierung von Bad stabilisiert den Komplex aus Bad und 14-3-3-Protein und schützt Neuronen vor dem Zelltod. Die Phosphorylierung von Bad erfolgt durch Proteinkinasen, zum Beispiel PKA und B/Akt, während ihre Dephosphorylierung durch die Phosphatasen Calcineurin oder durch die Proteinphosphatase 2C (PP2C) vermittelt wird (P. Chan, Stanford).
Eine Dephosphorylierung von Bad konnte auch beim apoptotischen Zelltod von Gefäßendothelzellen nachgewiesen werden. Dort wurde die Apoptose durch Inkubation mit solchen Fettsäuren ausgelöst, die auch eine Aktivierung der PP2C induzieren. Demnach könnte die Aktivierung von Bad auch an der (fettsäure-vermittelten) Schädigung des Gefäßendothels im Rahmen atherosklerotischer Prozesse beteiligt sein. Weitere Untersuchungen werden die spannende Frage klären, ob eine PP2C-Blockade eine wirksame therapeutische Strategie bei Atherosklerose ist (J. Schäfer, Marburg).
Folgen der gestörten Mitochondrienfunktion
Die Schädigung der Mitochondrien führt u. a. zu einem Abfall der zellulären Energiereserven und zur Freisetzung mitochondrialer Apoptosefaktoren wie Cytochrom C, Smac/Diablo und Omi, die im Zytosol die Aktivierung von Caspasen entscheidend verstärken. Ein anderes mitochondriales Protein, der Apoptose-induzierende Faktor (AIF), wird innerhalb der ersten Stunden nach der Ischämie (mehrere Stunden vor der Freisetzung von Cytochrom C) freigesetzt und gelangt rasch in den Zellkern. Dort vermittelt AIF die Zerstörung der DNA und leitet so – unabhängig von den Caspasen – den Zelltod ein (Abb. 2).
In kultivierten Neuronen und in entsprechenden Versuchen mit transgenen Tieren führte eine Reduktion von AIF um etwa 80% zu einer Abnahme der neuronalen Schädigung um ca. 50%. Die weitere Erforschung der Mechanismen der AIF-vermittelten Apoptose erscheint somit vielversprechend (V. Dawson, Baltimore; K. Blomgren, Göteborg; N. Plesnila, München).
Regeneration durch Aktivierung endogener Schutzmechanismen
Ebenso hoffnungsvoll sind neue Erkenntnisse über die Aktivierung von endogenen Schutzmechanismen, die das Überleben der Neuronen sichern können. Auch bei diesen Prozessen spielt die Phosphorylierung von Proteinen eine bedeutende Rolle. So hängt die neuroprotektive Wirkung des basischen Fibroblasten-Wachstumsfaktors (bFGF) von seiner Phosphorylierung im Extrazellularraum ab, denn der nicht phosphorylierte Wachstumsfaktor bleibt wirkungslos (S. Klumpp, Münster). In Hinsicht auf dieses Target könnte die Blockade von Phosphatasen mit niedermolekularen, gehirngängigen Substanzen eine neue Strategie für eine Schlaganfalltherapie sein.
Wachstumsfaktoren selbst stehen als Therapeutika nicht zur Verfügung, weil sie als Proteine nicht ohne Weiteres vom Blut in das Gehirngewebe gelangen können. Es kommt also darauf an, ihre endogene Synthese im Gehirngewebe durch niedermolekulare Substanzen, die die Blut-Hirn-Schranke leicht überwinden, zu steigern. Eine hierzu geeignete Substanz ist z. B. Clenbuterol, das allerdings bislang in experimentellen Schlaganfallmodellen mehrere Stunden vor der Schädigung gegeben werden musste, um eine neuroprotektive Wirkung zu entfalten.
Durch Kombination von Clenbuterol mit dem Glutamat-Rezeptorantagonisten Memantine ist es jetzt gelungen, das therapeutische Fenster von Clenbuterol auf mehrere Stunden nach Ischämie auszudehnen. Zudem war die Kombination der beiden Substanzen im Hinblick auf die Reduktion des Infarktvolumens stets wirksamer als die Einzelsubstanzen (C. Culmsee, München).
Epoetin, Statine und Sildenafil wirken neuroprotektiv
Ein Wirkstoff, der nicht nur in verschiedenen experimentellen Schlaganfallmodellen vielversprechende Wirkungen zeigte, sondern auch in ersten klinischen Studien an Schlaganfallpatienten erfolgreich eingesetzt wurde, ist das Erythropoietin (Epoetin). Es stimuliert über Wachstumsfaktoren wie VEGF und BDNF die Neubildung von Blutgefäßen (Angiogenese) und von Neuronen (Neurogenese). Besonders beeindruckend ist dabei, dass die Verbesserung von neurologischen Funktionen auch dann noch nachweisbar war, wenn der Wirkstoff bis zu 24 h nach dem ischämischen Insult appliziert wurde (M. Chopp, Detroit).
CEPO, ein Carbamyl-Derivat des Epoetins, besitzt offenbar die gleichen neuroprotektiven Eigenschaften wie Epoetin, ohne dass es die Hämatopoese beeinflusst. Während Epoetin durch eine gesteigerte Bildung von Retikulozyten, Leukozyten und insbesondere Thrombozyten das Thromboserisiko erhöht, waren nach Applikation von CEPO solche ungünstigen Blutbildveränderungen nicht nachweisbar (M. Leist, Valby).
Auch Cholesterolsyntheseenzym-Hemmer (Statine) und Phosphodiesterase-5-Inhibitoren (z. B. Sildenafil) verstärken die Angiogenese und Neurogenese nach einem Infarkt. Diese Wirkstoffe verbesserten neurologische Funktionen auch dann noch signifikant, wenn sie 24 h nach Ischämie gegeben wurden.
Akuttherapie des Schlaganfalls mit tPA ...
Obwohl in den vergangenen Jahrzehnten immer wieder neue Substanzen mit neuroprotektiven bzw. neuroregenerativen Wirkungen experimentell untersucht worden sind, stehen in der Klinik praktisch keine ausreichend wirksamen Arzneimittel zum breiten Einsatz bei Schlaganfallpatienten zur Verfügung. Viele der im Tiermodell vielversprechenden Ansätze waren am Schlaganfallpatienten nur eingeschränkt wirksam; das lag teilweise daran, dass die Arzneistoffe in den klinischen Studien viel zu spät oder zu niedrig dosiert eingesetzt wurden.
Die einzige derzeit verfügbare Therapie des Schlaganfalls liegt in der Verabreichung des Plasminogenaktivators tPA (Alteplase, Reteplase), der innerhalb von 3 h nach einem Gefäßverschluss gegeben werden muss, um ein Blutgerinnsel aufzulösen und das verschlossene Gefäß wieder zu öffnen. Eine mögliche Nebenwirkung dieser Therapie (insbesondere bei einer Applikation nach 3 h) liegt allerdings in der Auslösung von Blutungen im Gehirn, die dann fatale Folgen haben. Aufgrund dieses Risikos kann tPA nur bei etwa 4 bis 5% der Schlaganfallpatienten eingesetzt werden.
Nach neuen Ergebnissen führt tPA offenbar über die Aktivierung von Matrixmetalloproteinasen (MMP) zur erhöhten Blutungsneigung im Infarktgebiet. Möglicherweise könnten daher Kombinationen von tPA mit MMP-Inhibitoren die gefürchteten Nebenwirkungen deutlich reduzieren und damit die Lyse-Therapie auch zu späteren Zeitpunkten sicherer und effizienter machen (E. Lo, Charlestown; G. del Zoppo, La Jolla).
... oder mit Albumin und Docosanoiden
Ein anderer vielversprechender Wirkstoff, der bereits klinisch geprüft wird, ist Albumin. Es hat einen Anteil von 55 bis 62% an den Plasmaproteinen und zeigte in verschiedenen Schlaganfallmodellen eine ausgeprägte neuroprotektive Wirkung. Als Nebenwirkung der Albumintherapie traten bei einigen Patienten Störungen der Lungenfunktion auf. Inzwischen konnte die Albumindosis durch Kombination mit Docosanoiden deutlich reduziert werden, und klinische Studien der Phasen II und III sind in Vorbereitung (M. Ginsberg, Miami).
Docosanoide sind Derivate der Docosahexaensäure, die bei der oxidativen Spaltung von Membranlipiden nach einer zerebralen Ischämie entstehen und ausgeprägte neuroprotektive Eigenschaften besitzen. Das Docosanoid 10,17S-Docosatrien verminderte im Tiermodell die Einwanderung von Leukozyten und die Synthese von Entzündungsmediatoren wie z. B. COX-2 und NFκB und reduzierte das Infarktvolumen um fast 50% (N. Bazan, New Orleans).
In Zukunft: Regeneration von Gehirngewebe
Eine wichtige Erkenntnis des Symposiums war, dass in der Schlaganfalltherapie neben den Strategien zum unmittelbaren Schutz der Nervenzellen insbesondere die Unterstützung der Regeneration von Gehirngewebe eine herausragende Bedeutung erlangen wird. Hier kommen teilweise Arzneistoffe zum Einsatz, die in anderen Indikationsgebieten bereits etabliert sind, wie CSE-Hemmer, PDE-5-Inhibitoren, Albumin, Epoetin, Memantine oder der β2-Adrenozeptoragonist Clenbuterol. Auch Stammzellen könnten in Zukunft zu diesem Zweck therapeutisch eingesetzt werden.
Dank an Prof. Krieglstein
Seit 1986 hat Prof. Dr. Dr. Josef Krieglstein (Institut für Pharmakologie und Toxikologie, Fachbereich Pharmazie der Philipps-Universität Marburg) die internationalen Schlaganfallsymposien organisiert und alle zwei Jahre die führenden Spezialisten aus aller Welt in Marburg zusammengeführt. Mit der diesjährigen zehnten Tagung, auf der Wissenschaftler aus USA, Kanada, Japan, Korea, China, Indien, Israel, Saudi Arabien und ganz Europa rund 60 Vorträge und 90 Posterbeiträge präsentierten, endete diese Tradition.
Der Präsident der International Society for Cerebral Blood Flow and Metabolism, Prof. Iwao Kanno aus Japan, gratulierte Prof. Krieglstein zur erfolgreichen Durchführung der Schlaganfallsymposien und dankte im Namen aller Teilnehmer für die kontinuierlich hohe Qualität des international besetzten Programms. Als Zeichen des besonderen Danks wurde Prof. Krieglstein vom Beirat der Gesellschaft eine Ehrentafel mit folgendem Wortlaut überreicht: "The International Advisory Board recognizes Prof. Josef Krieglstein for his outstanding contributions to the neurosciences, for organizing the International Symposium on Pharmacology of Cerebral Ischemia during the last two decades and for academic leadership."
Prof. Traystman, Baltimore, meinte abschließend: "In Marburg wurden dank Prof. Krieglstein in einer bemerkenswert familiären Atmosphäre Generationen von Schlaganfallforschern über den neuesten Stand der Forschung unterrichtet, erfolgreiche Kollaborationen gebildet, neue Ideen entwickelt und so das Forschungsgebiet entscheidend vorangetrieben."
Danksagung:
Prof. Krieglstein dankte in seiner Begrüßungsansprache insbesondere der Deutschen Forschungsgemeinschaft, der International Society for Cerebral Blood Flow and Metabolism, der International Society for Neurochemistry und dem Fonds der Chemischen Industrie für die finanzielle Unterstützung des Symposiums.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.