

















- DAZ.online
- DAZ / AZ
- DAZ 38/2015
- Ähnlich, gleich oder ...
Orphan Drugs
Ähnlich, gleich oder klinisch überlegen?
Das Similarity Assessment bei Orphan Drugs
Disclaimer
Die im Folgenden geäußerten Sachverhalte sind persönliche Ansichten der Verfasser. Es handelt sich nicht um Ansichten des Bundesinstituts für Arzneimittel und Medizinprodukte, der European Medicines Agency oder eines ihrer Gremien oder Ausschüsse. Ein Rechtsanspruch entsteht nicht.
Im Lauf der letzten Jahre zeigte sich, dass die Ära der Blockbuster-Medikamente langsam ihrem Ende entgegen geht. Derzeitige Arzneimittelentwicklungs-Programme folgen oft dem Trend zu immer weitergehenden Stratifizierungen des Patientenkollektivs. Auf Basis von Mustern im Pharmakogenom und im Proteom werden immer kleinere Untergruppen für eine zielgerichtete Pharmakotherapie identifiziert. Die personalisierte Medizin kann als Biomarker-basierte Stratifizierung des Patientenkollektivs bis hinunter zum einzelnen Patienten betrachtet werden. Je exakter die Stratifizierung wird, desto seltener werden die Fälle, in denen ein Patient tatsächlich von einem Arzneimittel profitiert. Viele Arzneimittel-Neuentwicklungen richten sich entweder genuin gegen seltene Leiden (Orphan Diseases) oder erreichen in einer „häufigen“ Erkrankung nur Subpopulationen, die der Definition „selten“ entsprechen. Es verwundert daher nicht, dass die Zahl der Anträge auf Orphan Designation zunehmen. Orphan Drugs sind Arzneimittel, die gegen seltene Erkrankungen gerichtet sind, dies entspricht laut Definition der EMA einer Prävalenz von 5/10.000 Personen [European Medicines Agency 2015a]. Diese geringe Zielpopulation verringert die Gewinnmöglichkeit eines solchen Arzneimittelentwicklungs-Programms. Um Anreize für die Entwicklung von Orphan Drugs zu schaffen, wurde im Dezember 1999 die Verordnung EG/141/2000 verabschiedet, die Orphan Regulation [Europäisches Parlament 16. Dezember 1999]. Sie regelt unter anderem die geldwerten Anreize für die Entwicklung von Orphan Drugs und definiert die Voraussetzungen für eine Orphan Designation – ausführlicher lesen Sie dazu im Beitrag „Hilfe bei seltenen Krankheiten – Eine Einführung in die Orphan Drugs“ auf S. 58 in dieser DAZ. Des Weiteren ist in der Orphan Regulation geregelt, dass ein Arzneimittel gegen seltene Erkrankungen einen wesentlichen Beitrag zur Patientenversorgung (Major Contribution to Patient Care) erbringen muss und ein Significant Benefit erreicht werden kann. Hier setzen die wirtschaftlichen Anreizsysteme der europäischen Orphan-Gesetzgebung an. Attraktiv erscheint eine Orphan Designation nicht nur durch die wirtschaftlichen Vorteile, sondern auch durch weitere direkte und indirekte Unterstützungen seitens des Gesetzgebers. Generell werden Orphan Drugs im dem zentralen pan-europäischen Zulassungsverfahren zugeführt. Für Orphan Drugs kann unter bestimmten Voraussetzungen auch bei nicht ausreichender klinischer Datenlage eine Marktzulassung erreicht werden – als sogenannte Zulassung unter bestimmten Voraussetzungen (Marketing Authorisation under Exceptional Circumstances). Wenn die Regelung der Marktexklusivität ohne Ausnahmeregelungen angewendet würde, wären zehn Jahre Indikationsschutz für das erste Arzneimittel, welches in einer bestimmten Indikation als Orphan Drug zugelassen wurde, garantiert. Dies hätte zur Folge, dass der Fortschritt in der medizinischen Wissenschaft behindert würde. Dies ist aber nicht die Intention des Gesetzgebers: um einen Ausgleich zwischen Forschungsfortschritt und gerechter Behandlung der Unternehmen zu schaffen, wurde die Regelung der Orphan Similarity eingeführt. So kann ein weiteres Arzneimittel in einer bestehenden Indikation unter bestimmten Umständen selbst dann zugelassen werden, wenn eine Marktexklusivität vorliegt.
Ein zugelassenes Orphan Drug hat keinen Anspruch auf Marktexklusivität gegenüber einem weiteren Wirkstoff, falls dieser einen signifikanten Vorteil für die Behandlung bietet. Es handelt sich also um die Abwägung zweier Rechtsgüter: hier steht das Patientenwohl über dem vormals erworbenen Recht auf Marktexklusivität [European Medicines Agency 2015a]. Fazit: Die Marktexklusivität bietet dem pharmazeutischen Unternehmer nur einen relativen Schutz der Indikation, um die Patienten mit Arzneimitteln als zeitlich befristeter Monopolist versorgen zu können [vfa 2014].
Orphan Similarity
Der Begriff der „Similarity“ ist mehrfach besetzt im Rahmen arzneimittelrechtlicher Verfahren, daher ist stets ein spezifizierender Zusatz nötig, welche Art der Similarity im Einzelfall gemeint ist. Beispielsweise beschreibt die Essential Similarity generischer Produkte einen völlig anderen Zusammenhang, als die hier eine Rolle spielende Orphan Similarity.
In der Vergangenheit gab es verschiedene Fälle, bei denen mehrere Arzneimittel für die Behandlung einer ähnlichen bzw. überlappenden Indikation zugelassen wurden. Diese wurden mithilfe des Similarity Assessment bewertet. In der Tabelle 1 sind Beispiele dargestellt, bei denen trotz bestehender Marktexklusivität mehrere Präparate für eine Indikation zugelassen wurden – das Similarity Assessment bietet hierbei verschiedene Möglichkeiten [Dormeyer 2008].
Indikation |
Arzneimittel |
|---|---|
Behandlungen der pulmonalen Hypertonie |
|
multiples Myelom |
|
Philadelphia-Chromosom-positive chronisch-myeloische Leukämie (CML) |
|
Morbus Fabry |
|
Nierenzellkarzinom |
|
Wenn ein Produkt bereits als Orphan Drug zugelassen wurde und für einen weiteren Wirkstoff ein Antrag als Orphan Drug mit derselben Indikation gestellt wird, wird ein Similarity Assessment von den Experten der EMA (wissenschaftliche Assessoren der List of Experts) durchgeführt. Das Similarity Assessment betrachtet den Vergleich zweier Arzneistoffe (Active Pharmaceutical Ingredients, API) aus drei unterschiedlichen Perspektiven:
- aus der klinischen Perspektive, aus der die therapeutische Indikation geprüft wird,
- aus der präklinisch-pharmakologischen Perspektive, aus der der molekulare Wirkmechanismus untersucht wird, und
- aus der pharmazeutischen Perspektive, aus der die chemische Struktur betrachtet wird [European Commission 2008].
Im Similarity Assessment werden immer diese drei Eigenschaften beurteilt, obwohl bereits ein Unterschied in einer Eigenschaft ausreicht, um die Ähnlichkeit zu verneinen. Nur wenn alle drei Aspekte mit denen des bereits auf dem Markt befindlichen Arzneimittels übereinstimmen, besteht Similarity zwischen den geprüften Arzneimitteln. Dies hätte zur Folge, dass die Ähnlichkeitsprüfung positiv beschieden wird, es liegt also eine Orphan Similarity vor, durch die eine Zulassung zunächst ausgeschlossen wird. Damit das neue Arzneimittel mit derselben Indikation trotzdem auf den Markt darf, gibt es drei Ausnahmeregelungen [European Commission 2008].
- Eine Möglichkeit besteht im sogenannten Consent of MAH. Dabei gewährt der Zulassungsinhaber (Marketing Authorisation Holder, MAH) trotz bestehender Marktexklusivität dem Konkurrenzprodukt mit derselben Indikation die ausdrückliche Erlaubnis.
- Eine weitere Möglichkeit besteht im Nachweis des Insufficient Supply. Hierbei wird ein Versorgungsengpass (Drug Shortage) identifiziert und daher ein Antrag auf Zulassung befürwortet.
- Die dritte Möglichkeit besteht im Beleg der klinischen Überlegenheit (Clinical Superiority). Dies wiederum erfolgt durch den Nachweis in einer der drei folgenden Kategorien:
1. Nachweis einer besseren Wirksamkeit.
2. Nachweis eines besseren Sicherheitsprofils.
3. Nachweis eines wesentlichen Beitrags zur Patientenversorgung (Major Contribution to Patient Care).
Das Beispiel Imatinib und Nilotinib
Soll ein neues Orphan Drug für die gleiche Indikation zugelassen werden, wie ein bereits zugelassenes Arzneimittel für seltene Leiden, so muss eine Ähnlichkeitsprüfung durchgeführt werden. Diese erfolgte auch im Falle von Nilotinib (Tasigna®). Geprüft wurde 2006 die Ähnlichkeit von Nilotinib gegenüber Imatinib (Glivec®), das bereits 2001 als Tyrosinkinase-Inhibitor zugelassen worden war. Die Indikation für beide Substanzen lautete chronisch myeloische Leukämie (Chronic Myeloic Leucemia, CML). Im Folgenden wird das Similarity Assessment beispielhaft erläutert, das Imatinib und Nilotinib vergleichend untersucht hat.
Struktur der Moleküle
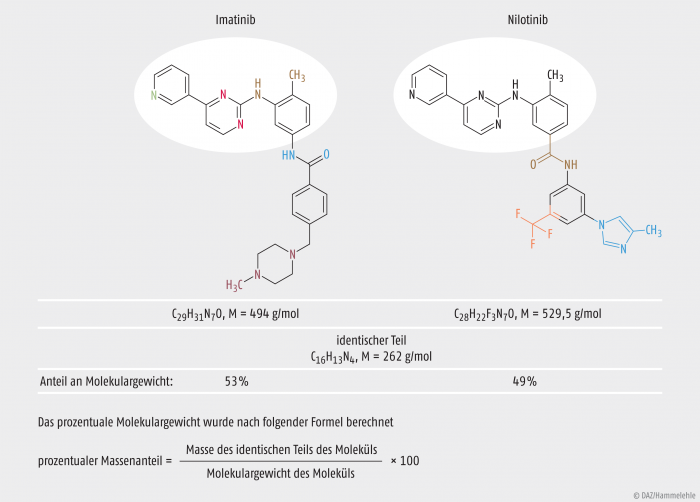
Die Struktur der beiden Wirkstoffe ist in der Abbildung 1 dargestellt. Zu erkennen ist der identische Teil der Struktur des Pharmakophors (weiß unterlegt). Das Grundgerüst der Strukturen besteht aus fünf heterocyclischen Verbindungen. Der identische Teil des Pharmakophors besteht aus einem Pyridinring (grün), welcher mit einem Pyrimidinring (rot) verbunden ist. Diese sind über die Aminogruppe des Methylanilins (braun) verbunden. Diese drei cyclischen Verbindungen bilden den identischen Teil der beiden Wirkstoffe. Bei Imatinib ist das Methylanilin über eine Amidkopplung an ein Benzamid (dunkelblau) gekoppelt, welches in para–Position mit einem Piperazin (pink) verbunden ist. Das Methylanilin des Nilotinib besitzt eine zusätzliche funktionelle Gruppe. Durch das Vorhandensein eines Amids handelt es sich im eigentlichen Sinne um ein Aminomethylbenzamid (braun). Dieses koppelt, durch die Amidkopplung, ein Trifluormethylphenyl (orange), an welchem wiederum ein Methylimidazol (hellblau) gebunden ist. Unterschiede bestehen also in den Gruppen des Methylanilins sowie in den nachfolgenden cyclischen Verbindungen. Diese Unterschiede wirken sich auf klinische und präklinische Parameter aus (Affinitäten, intrinsische Aktivitäten). Imatinib hat einen IC50-Wert von 600 nM am BCR-ABL-Rezeptor, Nilotinib gerade einmal 30 nM und besitzt somit eine höhere Bindungsaffinität zum Target [Selleckchem 2015]. Die erhöhte Targetaffinität ist auf die Substitution mit dem Trifluormethylphenyl und dem Methylimidazol zurückzuführen. Der Daylight-Fingerprint-Tanimoto-Koeffizient beträgt 0,6. Dieser Faktor dient dazu, chemische Strukturen zu vergleichen [Manley et al. 2010]. Verschiedene Software-Programme dienen der zwei- bzw. drei-dimensionalen Analyse der Molekülstrukturen [European Commission 2008], um diese besser miteinander vergleichen zu können. Bei der Ähnlichkeitsprüfung wurde den Wirkstoffen Ähnlichkeit bescheinigt: Imatinib und Nilotinib sind „Orphan Similar“ zueinander. Der CHMP verglich das prozentuale Molekulargewicht (mindestens 50% identisch) der übereinstimmenden Partialstruktur [Manley et al. 2010] und bezog weitere Parameter der Molekülähnlichkeit in die Entscheidungsfindung ein.
Abb. 1: Imatinib vs. Nilotinib – Ähnlichkeit der chemischen Struktur
Die therapeutischen Indikationen
Imatinib und Nilotinib werden unter den Handelsnamen Glivec® bzw. Tasigna® vertrieben. Ein weiterer Punkt des Similarity Assessments umfasst die Indikationen. Beide Arzneimittel unterschieden sich zunächst, da Nilotinib als Derivat von Imatinib nur als Zweit-Linien-Therapie für die CML vorgesehen war. Zum Zeitpunkt der Einreichung des Zulassungsantrags für Nilotinib (Tasigna®) lauteten die Indikationen: „Für die Behandlung von Erwachsenen mit Philadelphia-Chromosom positiver (Ph+) chronischer myeloischer Leukämie (CML) in der chronischen und akzelerierten Phase mit Resistenz oder Unverträglichkeit gegenüber einer Vorbehandlung einschließlich Imatinib. Wirksamkeitsdaten zu Patienten mit CML in der Blastenkrise liegen nicht vor.“
Die Erweiterung der Indikation von Nilotinib erfolgte im Zuge einer Variation und wurde 2010 akzeptiert [Committee for Medicinal Products for Human Use 2010]. Somit ist Nilotinib also auch als Erst-Linien-Therapie zur Behandlung von CML-Patienten vorgesehen [European Medicines Agency 2015b]. Imatinib (Glivec®) wurde 2001 zugelassen für die Indikationen
- Behandlung der chronisch-myeloischen Leukämie und
- Behandlung von gastrointestinalen Stromatumoren.
2005 wurde das Profil erweitert um fünf zusätzliche Indikationen:
- Ph+CML in der chronischen Phase nach Versagen einer Interferon-Alpha-Therapie, in der akzelerierten Phase oder in der Blastenkrise,
- Erwachsene und Kinder mit neu diagnostizierter Philadelphia-Chromosom-positiver akuter lymphatischer Leukämie (Ph+ALL) in Kombination mit einer Chemotherapie,
- Erwachsene mit rezidivierter oder refraktärer Ph+ALL als Monotherapie,
- Erwachsene mit myelodysplastischen/myeloproliferativen Erkrankungen (MDS/MPD) in Verbindung mit Genumlagerungen des PDGF-Rezeptors (platelet-derived growth factor),
- Erwachsene mit fortgeschrittenem hypereosinophilem Syndrom (HES) und/oder chronischer eosinophiler Leukämie (CEL) mit FIP1L1-PDGFRα-Umlagerung [Novartis Pharma 2014].
Somit ergab das Similarity Assessment keine direkte Übereinstimmung der Indikationen, wie die Tabelle 2 zeigt [European Medicines Agency 2012a].
Imatinib |
Nilotinib |
|---|---|
|
Behandlung von Erwachsenen und Kindern mit neu diagnostizierter Philadelphia-Chromosom positiver chronischer myeloischer Leukämie (CML), in der chronischen und akzelerierten Phase, für die eine Knochenmarktransplantation als Erstbehandlungsmöglichkeit nicht in Betracht gezogen wird.
|
Behandlung von Erwachsenen mit Philadelphia-Chromosom positiver chronischer myeloischer Leukämie (CML) in der chronischen und akzelerierten Phase mit Resistenz oder Unverträglichkeit gegenüber einer Vorbehandlung einschließlich Imatinib.
|
Der molekulare Wirkmechanismus
Beide Moleküle besitzen zu einem großen Anteil identische Zielstrukturen. Nilotinib inhibiert wie Imatinib die Rezeptoren Kit und PDGF (platelet-derived growth factor), sowie die BCR-ABL-Tyrosinkinase. Unterschiede finden sich in Interaktionen mit drei weiteren Rezeptoren (DDR; CSF; c-Src) [European Medicines Agency (2012a) und (2013)]. Für die Indikation der Wirkstoffe und die Behandlung der CML ist die Inhibition des BCR-ABL-Rezeptors entscheidend. Da beide Wirkstoffe in der CML-Therapie am gleichen Wirkort inhibierend angreifen, besteht auch beim Wirkmechanismus durchaus eine Ähnlichkeit, da der Wirkmechanismus zweier Arzneimittel, die mehrere Targets besitzen, als ähnlich betrachtet wird, wenn die primäre pharmakodynamische Wirkung beider Arzneimittel identisch ist [European Commission 2008, Eckstein et al. 2014].
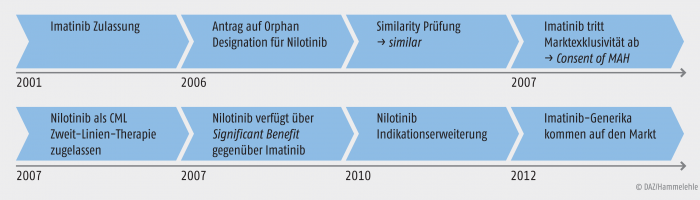
Die Wirkmechanismen sind besonders interessant, da die Therapie der CML durch Imatinib revolutioniert wurde: bevor Imatinib als erster Tyrosinkinase-Inhibitor zur Therapie der CML zugelassen wurde, lag die Fünf-Jahres-Überlebensrate bei ungefähr 20%. Unter Imatinib verbesserte sie sich auf über 90% [O‘Brien et al. 2003]. Dies weist auf die Vorteile einer zielgerichteten Therapie hin [Quintás-Cardama und Cortes 2006]. Die Variation der Molekülstruktur zwischen Imatinib und Nilotinib ermöglichte es, Nilotinib als Zweit-Linien-Therapie zuzulassen, da Resistenzmechanismen umgangen werden können [Giles 2010]. Zusammengenommen besitzt Nilotinib also eine Orphan Similarity zu Imatinib, welche eine Zulassung im gleichen Indikationsgebiet ausschließen sollte. Jedoch kann mit dem Einverständnis des Zulassungsinhabers von Imatinib eine Zulassung im Indikationsgebiet genehmigt werden. Da im Falle von Imatinib und Nilotinib der Zulassungsinhaber identisch war, wurde die Zulassung ermöglicht [Europäische Kommission 2007b]. Für Tasigna® (Nilotinib) wurde nach der Zulassung durch die Europäische Kommission vom COMP ein Significant Benefit anerkannt und damit der Orphan Drug Status mit der damit verbundenen Marktexklusivität gewährt. Die zu diesem Zeitpunkt noch drei Jahre bestehende Marktexklusivität von Imatinib gegenüber dritten Arzneimitteln blieb davon unberührt.
Glossar
Es ist derzeit geplant eine harmonisierte Darstellung ausgewählter Begriffe im Zusammenhang mit dem Similarity Assessment unter Federführung der EMA festzulegen. Für zukünftige regulatorische Verfahren werden Begriffe wie Benefit oder Superiority bestimmt, um Uneinigkeiten zu vermeiden. Bis zur endgültigen Charakterisierung werden folgende Begriffe verwendet:
Significant Benefit: EG/141/2000
Es handelt sich hierbei um einen relevanten klinischen Nutzen mit hoher Wahrscheinlichkeit, dass der Patient tatsächlich diesen Nutzen spürt oder bemerkt. Ein Significant Benefit gilt immer im Vergleich zur aktuellen Therapiemöglichkeit und wird vom COMP geprüft. Als wichtiger Indikator dient vor allem die Evidenz der vorgelegten Daten.
Beispiele:
- Vorteile für eine bestimmte Patientengruppe, einschließlich resistenter Patienten gegenüber einer bestehenden Methode.
- Ein klinisch relevantes verbessertes Sicherheitsprofil.
- Eine verbesserte Formulierung oder Applikation, falls ernsthafte und dokumentierte Schwierigkeiten mit der Formulierung oder Art der Verabreichung bestehen.
Market Exclusivity EG/141/2000 und EG/847/2000
Es wird keine Marktzulassung für Similar Medicinal Products innerhalb der gleichen Indikation erteilt, solange eine Marktexklusivität besteht.
„3.(c) … der zweite Antragsteller kann jedoch im Antrag des Arzneimittels, welches auch similar zum ersten Arzneimittel sein kann, darlegen, dass sein Arzneimittel sicherer, effektiver oder auf eine andere Weise klinisch Überlegen ist ...“
Cinical Superiority: EG/847/2000
„3.(d) … ‚klinisch überlegen‘ bedeutet, dass ein Arzneimittel im Vergleich zu einem zugelassenen Arzneimittel für seltene Leiden nachweislich zusätzlich einen oder mehrere der im Folgenden genannten erheblichen therapeutischen Vorteile aufweist: [...]“
- größere Wirksamkeit,
- größere Sicherheit oder
- in außergewöhnlichen Fällen ist der Nachweis zu erbringen, dass das Arzneimittel einen bedeutenden Beitrag zur Diagnose oder Behandlung von Patienten leistet.
Significant Clinical Benefit: EG/726/2004
Dieser Begriff wird im Zusammenhang mit einer Indikationserweiterung, innerhalb der ersten acht Jahre der zehnjährigen Marktexklusivität, benutzt. Das Orphan Drug muss einen Significant Clinical Benefit gegenüber der gängigen Therapie aufweisen, um ein weiteres Jahr Marktexklusivität zu sichern.
Major Contribution to Patient Care: EG/141/2000
Die signifikante Verbesserung der Patientenversorgung basiert auf einer Verbesserung der Arzneimittelverträglichkeit, einer optimierten galenischen Formulierung oder einer erweiterten Verfügbarkeit des Arzneimittels für das Patientenkollektiv. Eine weitere Möglichkeit ein Major Contribution to Patient Care zu erzielen, besteht darin, die Lebensqualität der Patienten zu verbessern.
Orphan Similarity
Ein Orphan Drug gilt als similar zu einem weiteren Orphan der selben Indikation, wenn einer der Punkte Struktur, Wirkmechanismus oder Indikation übereinstimmen, während einer aktiven Marktexklusivität.
Imatinib als „regulatorisches Gambit“?
Das ursprünglich aus dem Italienischen stammende Wort Gambit bezeichnet einen Vorgang beim Schachspiel: eine Figur wird für einen strategischen Vorteil oder einen Stellungsvorteil geopfert. Wurde Imatinib also ein Opfer der Marktexklusivität von Nilotinib?
Imatinib (Glivec®) wurde 2001 als Orphan Drug zentral zugelassen. Als Orphan Drug hatte es einen Anspruch auf eine zehnjährige Marktexklusivität für die zugelassene Indikation der chronischen myeloischen Leukämie (CML) bei Erwachsenen als Erst-Linien-Therapie. Das Nachfolgepräparat Nilotinib wurde etwa sieben Jahre nach Zulassung von Imatinib ebenfalls zur Zulassung eingereicht und als „similar“ zu Imatinib bewertet. Da beide Arzneimittel vom selben Unternehmen entwickelt wurden, wurde der Konflikt der angestrebten Zulassung von Nilotinib mit der Marktexklusivität von Imatinib durch das Einverständnis des Inhabers der Imatinib-Zulassung mit der Zulassung von Nilotinib aufgelöst.
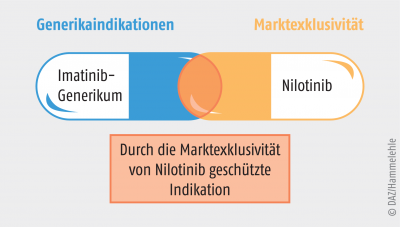
Dies erscheint auf den ersten Blick als ein Nachteil für Nilotinib, da mit der Feststellung der Similarity seine innovative Bedeutung geschwächt wirkte. Nach der Zulassung von Nilotinib durch die Europäische Kommission wurde vom COMP der Significant Benefit für Nilotinib und dessen Orphan-Drug-Status bestätigt. Damit begann zum Zeitpunkt der Nilotinib-Zulassung dessen eigener zehnjähriger Anspruch auf Marktexklusivität. Mit dem Ende der Marktexklusivität von Imatinib wären prinzipiell Imatinib-haltige Generika zum Original Glivec® möglich gewesen, die alle Indikationen von Imatinib beanspruchen können. Allerdings ergab sich für diese Generika ein Konflikt mit der Marktexklusivität von Nilotinib. Es war bereits bei der Zulassung von Nilotinib entschieden worden, dass Imatinib und Nilotinib als similar zu betrachten sind. Für alle von Nilotinib beanspruchten Indikationen ist damit eine Zulassung von Generika mit dem Wirkstoff Imatinib ausgeschlossen, solange die Marktexklusivität von Nilotinib besteht. In anderen Worten, für zehn Jahre nach der Zulassung von Nilotinib können keine Imatinib-Generika zugelassen werden, die Indikationen von Nilotinib beanspruchen. Generische Arzneimittel mit dem Wirkstoff Imatinib können daher nicht wie üblich alle Indikationen des Referenz-Arzneimittel Glivec® abbilden, sondern sind nur mit den Indikationen möglich, die zwar für Glivec® (Imatinib), aber nicht auch für Tasigna® Nilotinib bestehen.
Dies ist auch der Grund, weshalb die Indikationen des Imatinib-Originators Glivec® und der Imatinib-Generika nicht identisch sind (was man bei einem „normalen“ Generikum erwarten würde). Ersichtlich wird dies aus Abbildung 2.
Abb. 2: Schnittmenge der Indikationen
Abbildung 3 verdeutlicht die Zusammenhänge des Ablaufes, rund um die Nilotinib-Marktexklusivität. Das erste generische Produkt von Glivec®, Imatinib-Teva®, erschien im Oktober 2012 auf dem Markt. Nichtsdestotrotz bleiben Imatinib noch die Überschneidungen mit Nilotinib, welche nicht von generischen Arzneimitteln abgedeckt sind, wobei in diesem Fall ein Off-label-use nicht auszuschließen ist [Committee for Medicinal Products for Human Use 2012].
Abb. 3: Die Abläufe rund um Imatinib und die Marktexklusivität
Fazit und Ausblick
Die große Anzahl an Neuzulassungen im Bereich der Orphan Drugs hebt die Relevanz der europäischen Orphan-Gesetzgebung hervor. Seit Inkrafttreten der Verordnung EG/141/2000 sind aktuell 88 Arzneimittel als Orphan Drug zentral zugelassen und ca. 1200 haben eine Orphan Designation. Derzeit umfassen die zugelassenen Arzneimittel gegen seltene Leiden ungefähr 30 Indikationen [European Commission: Community Register, 17. März 2015]. Trotz der Anreize für die pharmazeutische Industrie, werden bei Weitem noch nicht alle Indikationen abgedeckt. Die Europäische Kommission geht davon aus, dass es 5000 bis 8000 seltene Leiden gibt [European Medicines Agency, 2012 b].
Die Vergabe der Marktexklusivität scheint auf den ersten Blick eine strenge und starre Gesetzesstruktur zu sein. Wie die oben angeführten Ausnahmen zeigen, ist dem nicht so. Mithilfe des Similarity Assessments wurde ein faires Gleichgewicht zwischen Schutz des Erstanbieters und Forschungsfortschritt etabliert. Zusammenfassend kann gesagt werden, dass die Problematik der europäischen Gesetzgebung zu den Arzneimitteln für seltene Erkrankungen darin liegt, die optimale Balance zwischen dem Schaffen von Anreizen und der Verhinderung des Missbrauchs der Gesetzgebung zu erreichen. |
Literatur
Committee for Medicinal Products for Human Use (2005): Guideline on Procedures for the granting of a marketing authorisation under exceptional circumstances, pursuant to Article 14 (8) of Regulation (EC) NO 726/2004 Hg. v. European Medicines Agency
Committee for Medicinal Products for Human Use (2010): Summary of opinion (post authorisation). Tasigna - Nilotinib. Hg. v. European Medicines Agency
Committee for Medicinal Products for Human Use (2012): Assessment report. Imatinib Teva. EMA/CHMP/593709/2012. Online verfügbar unter www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002585/WC500137417.pdf.
Dormeyer, M. (2008): Regulatory strategies and practical aspects for the development and authorisation of orphan medicinal products in the european union
Eckstein, N. Arzneimittel - Entwicklung und Zulassung. Für Studium und Praxis. Deutscher Apotheker Verlag Stuttgart, 2013
Eckstein N, Röper L, Haas B, Potthast H, Hermes U, Unkrig C et al. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. In: Journal of experimental & clinical cancer research 2014;CR33:15, DOI: 10.1186/1756-9966-33-15
Europäische Kommission (27. April 2000): Verordnung (EG) Nr. 847/2000 zur Festlegung von Bestimmungen für die Anwendung der Kriterien für die Ausweisung eines Arzneimittels als Arzneimittel für seltene Leiden und von Definitionen für die Begriffe „ähnliches Arzneimittel“ und „klinische Überlegenheit“. EG/847/2000
Europäische Kommission (2007a): Entscheidung der Kommission über die Genehmigung für das Inverkehrbringen des Humanarzneimittels für seltene Leiden „Tasigna - Nilotinib“ gemäß der Verordnung EG Nr. 726/2004 des Europäischen Parlaments und des Rates. Online verfügbar unter http://ec.europa.eu/health/documents/community-register/2007/2007111932888/dec_32888_de.pdf.
Europäische Kommission (2007b): Entscheidung der Kommission über die Genehmigung für das Inverkehrbringen des Humanarzneimittels für seltene Leiden „Tasigna - Nilotinib“ gemäß der Verordnung EG Nr. 726/2004 des Europäischen Parlaments und des Rates. Online verfügbar unter http://ec.europa.eu/health/documents/community-register/2007/2007111932888/dec_32888_de.pdf
Europäisches Parlament (16. Dezember 1999): Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates über Arzneimittel für seltene Leiden. Verordnung EG/141/2000
Europäisches Parlament (30. April2004): Verordnung (EG) Nr. 726/2004 zur Festlegung von Gemeinschaftsverfahren für die Genehmigung und Überwachung von Human und Tierarzneimitteln und zur Errichtung einer Europäischen Arzneimittel-Agentur. Artikel 14(8) EG/726/2004
European Commission: Pharmaceuticals - COMMUNITY REGISTER. Register of designated Orphan Medicinal Products (by number). Hg. v. European Commission, Zugriff am 17. März 2015
European Commission (2008): Guideline on aspects of the application of Article 8(1) and (3) of Regulation (EC) No 141/2000: Assessing similarity of medicinal products versus authorised orphan medicinal products benefiting from market exclusivity and applying derogations from that market exclusivity. Online verfügbar unter http://ec.europa.eu/health/files/orphanmp/doc/c_2008_4077_en.pdf.
European Medicines Agency (2012a): EPAR summary for the public. Tasigna - Nilotinib.
European Medicines Agency (2012b): Significant benefit of orphan drugs: concepts and future developments
European Medicines Agency (2013): EPAR summary for public. Glivec- Imatinb. European Medicines Agency
European Medicines Agency (2015a): Rare disease (orphan) designations. Hrsg. European Medicines Agency, zuletzt aktualisiert am 15. Januar 2015
European Medicines Agency (2015b): Tasigna - Nilotinib, zuletzt aktualisiert am 6. Februar 2015
G-BA (2013 a):Dossier zur Nutzenbewertung gemäß § 35a SGB V; Ponatinib (Iclusig®) Ariad Pharmaceuticals (Germany) GmbH, Modul 2, www.g-ba.de/downloads/92-975-342/2013-08-01_Modul2_Ponatinib.pdf
G-BA (2013 b): Erstellung und Einreichung eines Dossiers zur Nutzenbewertung gemäß § 35a SGB V. Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards
Giles, FJ, Rosti G, Beris P, Clark RE, Le Coutre P, Mahon F-Xavier et al. Nilotinib is superior to imatinib as first-line therapy of chronic myeloid leukemia: the ENESTnd study. In: Expert review of hematology 2010;3(6):665–673, DOI: 10.1586/ehm.10.61
Manley PW, Stiefl N, C-J, Sandra W, Kaufman S, Mestan J, Wartmann M et al. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. In: Bioorganic & medicinal chemistry 2010:18(19): 6977–6986. DOI: 10.1016/j.bmc.2010.08.026
Novartis Pharma (2014): Fachinformation Glivec® (Zusammenfassung der Merkmale des Arzneimittels/SmPC) O‘Brien SG, Guilhot F, Larson, R, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. In: N Engl J Med 2003;348(11):994–1004, DOI: 10.1056/NEJMoa022457
Quintás-Cardama A, Cortes J. Chronic myeloid leukemia: diagnosis and treatment. In: Mayo Clinic proceedings 2006; 81(7):973–988, DOI: 10.4065/81.7.973
Nilotinib. Selleckchem, 2015, www.selleckchem.com
Was der Orphan Drug-Status für ein Medikament bedeutet (und was nicht). Hrsg. Die forschenden Pharma Unternehmen (vfa), 2014
Autoren
Vladlena Pfeifer studiert pharmazeutische Chemie und verbringt die Praxisphase ihres Studiums bei Prof. Dr. Niels Eckstein im Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM).
Anja Metzger hat angewandte Pharmazie an der Hochschule Kaiserslautern studiert und schreibt ihre Abschlussarbeit bei Dr. Paesler und Prof. Eckstein am BfArM.
Martin Norta ist Apotheker, verfügt über viele Jahre Erfahrung im regulatorischen Bereich und leitet im BfArM das Fachgebiet 21 (Verfahrensmanagement/Drug Regulatory Affairs).
Priv.-Doz. Dr. Harald Enzmann ist habilitierter Mediziner und langjährig erfahrener Wissenschaftler im onkologischen Bereich. Er leitet die Abteilung 2 Zulassung im BfArM und ist Mitglied im Ausschuss für Humanarzneimittel der EMA (CHMP).
Prof. Dr. Niels Eckstein ist Wissenschaftler am Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und Professor für Regulatory Affairs und Pharmakologie am Campus Pirmasens der FH Kaiserslautern.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.