




















- DAZ.online
- DAZ / AZ
- DAZ 49/2004
- Wie werden Arzneimittel ...
Kongress
Wie werden Arzneimittel bewertet?
Wann ist ein Arzneimittel bedenklich?
Das deutsche Arzneimittelgesetz verbietet, bedenkliche Arzneimittel in den Verkehr zu bringen. Prof. Dr. Jürgen Beckmann vom Bundesinstitut für Arzneimittel und Medizinalprodukte (BfArM), Bonn, erläuterte den entsprechenden Paragraphen Schritt für Schritt.
Das Verbot, bedenkliche Arzneimittel in den Verkehr zu bringen (§ 5 Abs. 1), betrifft alle Fertigarzneimittel, aber auch Diagnostika, Rezepturarzneimittel, Prüfarzneimittel und Gewebetransplantate. Es gilt für jeden, der ein Arzneimittel in den Verkehr bringt. Das ist primär der pharmazeutische Unternehmer, es sind aber auch Apotheker (bei der Abgabe von Rezepturarzneimitteln), Ärzte (z.B. bei der Frischzellentherapie) sowie Sponsor und Prüfarzt bei klinischen Studien.
Bedenklich sind für den Gesetzgeber "Arzneimittel, bei denen nach dem jeweiligen Stand der wissenschaftlichen Erkenntnis der begründete Verdacht besteht, dass sie bei bestimmungsgemäßem Gebrauch schädliche Wirkungen haben, die über ein nach den Erkenntnissen der medizinischen Wissenschaft vertretbares Maß hinausgehen" (§ 5 Abs. 2 AMG).
Schädliche Wirkungen
Mit schädlichen Wirkungen sind im Wesentlichen unerwünschte Arzneimittelwirkungen (UAW) gemeint, bei denen
- Schweregrad (z.B. anaphylaktischer Schock als schwerwiegend, Flush als nicht schwerwiegend),
- Dauer und Reversibilität (z.B. Urtikaria als reversible, ein Karzinom als irreversible UAW),
- Erkennbarkeit und Vermeidbarkeit (z.B. eine Agranulozytose ist im Blutbild erkennbar, eine Resistenz gegen aktiviertes Protein-C nicht) sowie,
- die Häufigkeit (von sehr selten < 0,01% bis sehr häufig > 10%)
beurteilt werden (Tab. 1). Bei der Risikoeinschätzung ist es sinnvoll, zwischen kontinuierlichen und dichotomen unerwünschten Arzneimittelwirkungen zu unterscheiden. Erstere treten praktisch immer auf, können in jedem Schweregrad vorkommen und sind messbar. Demgegenüber sind dichotome Nebenwirkungen sehr selten, schwer wiegend und direkt zählbar. Häufig gehen kontinuierliche Nebenwirkungen den dichotomen voraus ("Vor dem Brechen kommt das Biegen").
Tab. 1: Typologie der UAW
| schwer wiegend | nicht schwer wiegend |
| tödlich lebensbedrohend zu einer Klinikeinweisung führend einen Klinikaufenthalt velängern zu anhaltender Behinderung führend zu kongenitaler Anomalie führend Beispiele: Agranulozytose, Lungenembolie | alles andere Beispiele: Schlafstörung, Haarausfall |
| TYP A | TYP B |
| typisch für pharmakologische Wirkung dosisabhängig vorhersehbar oft nicht schwer wiegend Beispiele: Tachykardie durch Adrenalin, Osteoporose durch Prednison | Hypersensitivität (meist allergische Reaktion) weit gehend dosisunabhängig kaum vorhersehbar meist schwer wiegend Beispiel: Lyell-Syndrom durch Sulfonamide |
| kontinuierlich | dichotom |
| häufig bis immer in jeder Schwere quantifizierbar Beispiele: Injetkionsschmerz, Osteoporose | eher selten immer gleich schwer definiert, zählbar Beispiele: Fraktur, Tod |
Begründeter Verdacht
Im Gegensatz zu strafrechtlichen Bedingungen, bei denen ein begründeter Verdacht nicht zu einer Verurteilung führen kann, reicht bei Arzneimitteln eine vermutete Bedenklichkeit aus, um Maßnahmen zu ergreifen. Das bedeutet, es muss noch kein definitiver Beweis erbracht sein, dass das Arzneimittel bedenklich ist. Die erforderliche Evidenz ist umso geringer, je höher das vermutete Risiko ist.
Bestimmungsgemäßer Gebrauch
Unter einem bestimmungsgemäßen Gebrauch ist in erster Linie die Anwendung gemäß der Gebrauchs- und Fachinformation gemeint. Nach gültiger Rechtssprechung ist aber auch eine Anwendung außerhalb dieses Rahmens "bestimmungsgemäß", wenn sie medizinisch anerkannt ist, de facto häufig praktiziert wird oder nahe liegt.
Vertretbarkeit
Das vertretbare Maß kann nur von Fall zu Fall definiert werden und unter Abwägen von Nutzen und Risiko erfolgen. Die Gefahr, die von einer Erkrankung ausgeht, muss mit den potenziellen unerwünschten Wirkungen eines Arzneimittels aufgerechnet werden. Ferner ist zu berücksichtigen, ob zur Therapie einer bestimmten Erkrankungen nur ein Arzneimittel zur Verfügung steht, wie dies zum Beispiel bei den ersten Therapieansätzen bei AIDS der Fall war, oder ob weitere therapeutische Alternativen bestehen. Das vertretbare Maß ist ein individuelles Maß, so kann für einen Piloten ein Medikament mit sedierenden Nebenwirkungen nicht akzeptabel sein, eine Schauspielerin wird ein Arzneimittel, das zu Haarausfall führt, ablehnen. Das heißt also, der Vertretbarkeit liegt immer eine Nutzen-Risiko-Konstellation zugrunde.
Typische Nutzen-Risiko-Konstellationen*
- Ein Arzneimittel zur Behandlung einer schweren Erkrankung mit hoher Letalitätsrate hat trotz Vorsichtsmaßnahmen gelegentlich tödliche UAW. Da das Arzneimittel wirksam ist, wird das akzeptiert.
- Bei einem Arzneimittel, das bei einer chronischen, stark beeinträchtigenden Krankheit die Lebensqualität nachweislich verbessert, wird ein gewisses Risiko toleriert.
- Bei prophylaktisch gegebenen Arzneimitteln wie Impfstoffen sind hohe Sicherheitsanforderungen erforderlich.
- Arzneimittel ohne nachgewiesenen Nutzen können schon bei Bekanntwerden weniger schwerwiegender UAW mit hinreichendem Kausalzusammenhang inakzeptabel sein.
- Bei zwei Arzneimitteln mit gleicher Wirksamkeit und gleichartigen UAW wird das mit der größeren UAW-Häufigkeit nicht akzeptiert.
* Laut EU Pharmacovigilance Working Party
Wer trägt die Beweislast?
Beim Zulassungsverfahren muss der pharmazeutische Unternehmer den Nutzen seines Medikaments nachweisen und in Beziehung zu dem der therapeutischen Alternativen stellen. Dass ein Risiko nicht existiert, lässt sich nicht beweisen, es kann nur anhand toxikologischer Daten und aussagekräftiger Studien gezeigt werden, dass das Risiko relativ gering ist. Der Antragsteller beweist also primär den Nutzen. Anders verhält es sich bei einem Widerruf der Zulassung im Stufenplanverfahren durch das BfArM. Hier wird das Risiko bewiesen.
Die Abwesenheit von Nutzen kann die Behörde nicht beweisen. Sie kann nur argumentieren, dass der gezeigte Nutzen im Verhältnis zum Risiko oder zu dem der therapeutischen Alternativen gering ist, oder dass die Studien zum beanspruchten Nutzen unzureichend sind.
Beurteilung der Wirksamkeit von Arzneimitteln
"Seitdem man begonnen hat, die einfachsten Behauptungen zu beweisen, erwiesen sich viele von ihnen als falsch". Mit diesem Zitat von Bertrand Russell begann Dr. Eva Susanne Dietrich von der Kassenärztlichen Bundesvereinigung in Berlin ihren Vortrag, in dem sie potenzielle Mängel von Studien und traditionellen Therapien aufzeigte und erläuterte, nach welchen Kriterien Studienaussagen zu bewerten sind.
Im Zuge der Kosteneinsparung und mit der Etablierung von Leitlinien werden Arzneimittel und Therapieformen neu bewertet. Dies führt zu einer kritischen Sichtung des traditionellen Vorgehens und mitunter zu einer völlig neuen Bewertung. Dietrich führte hierzu einige Beispiele auf: Seit langem wird Magnesium gegen Wadenkrämpfe eingenommen, obwohl keine evidenzbasierte Studie hierfür vorlag. Eine erst 1999 durchgeführte Untersuchung zeigte keinen Benefit für Magnesium bei Wadenkrämpfen. Ein weiteres Beispiel ist die meist unnötige Verordnung von Antibiotika bei Otitis media im Kindesalter. Rein rechnerisch müssen 17 Kinder mit Antibiotika therapiert werden, um bei einem Kind die Schmerzen zu verhindern. Der Nutzen einer solchen Therapie ist fragwürdig.
Das Studiendesign
Bereits das Studiendesign gibt Hinweise auf die Evidenz einer Studie (Tab. 2). Handelt es sich um eine randomisierte kontrollierte Studie (hoher Evidenzgrad) oder um eine Anwendungsbeobachtung (niederer Evidenzgrad)? Randomisierte Studien haben das überzeugendste Design und können kaum durch Bias (Verzerrung von Studienergebnissen durch systematische Fehler) beeinflusst werden. Sie sind das Mittel der Wahl bei Vergleichsuntersuchungen gegenüber einer Plazebotherapie bzw. anderen therapeutischen Standardverfahren. Sie sind teuer und in ihrer Konzeption und Ausführung mit hohem Aufwand verbunden. Anwendungsbeobachtungen, von denen in Deutschland pro Jahr rund 400 durchgeführt werden, sind praxisnah, einfach zu konzipieren und können Antwort auf viele unterschiedliche Fragen geben.
Tab. 2: Einteilung klinischer Studien nach ihrem Evidenztyp.
| Stufe | Evidenztyp |
|---|---|
| Ia | Evidenz aufgrund von Metaanalysen randomisierter, kontrollierter Studien |
| Ib | Evidenz aufgrund mindestens einer randomisierten, kontrollierten Studie |
| IIa | Evidenz aufgrund mindestens einer gut angelegten kontrollierten Studie ohne Randomisierung |
| IIb | Evidenz aufgrund mindestens einer gut angelegten quasi-experimentellen Studie (z.B. Kohortenstudie) |
| III | Evidenz aufgrund gut angelegter, nicht experimenteller deskriptiver Studien (z.B. Vergleichsstudien, Fall-Konstruktionen) |
| IV | Evidenz aufgrund von Einzelfallberichten, Meinungen von Expertenkreisen, Konsensuskonferenzen und / oder klinischer Erfahrung anerkannter Autoritäten |
Reviews und Metaanalysen werden zu den Sekundärstudien gezählt. Systematische Reviews liefern die besten und aussagekräftigsten Informationen im Bereich der Sekundärliteratur. Es sind wissenschaftliche Literaturübersichten, die Schlussfolgerungen auf therapeutische Entscheidungen zulassen. Metaanalysen sind die statistische Ergänzung der systematischen Betrachtung aller Untersuchungen, die weltweit zu einer bestimmten klinischen Fragestellung durchgeführt wurden. Wird eine Metaanalyse korrekt durchgeführt, besitzt das Ergebnis eine höhere Aussagekraft als die zugrunde liegenden Untersuchungen.
Wichtige Studienparameter
Ein aussagekräftiger Studienparameter ist die NNT (number needed to treat). Sie gibt die Zahl der Patienten an, die behandelt werden müssen, um ein zusätzliches positives Zielergebnis zu erreichen. Anhand eines Beispiels zeigte Dietrich auf, dass einzelne Studienparameter einen positiven Eindruck erwecken, wird aber die NNT ermittelt, findet sich oftmals ein enttäuschendes Ergebnis.
Der p-Wert (probability of error) gibt die Wahrscheinlichkeit an, mit der ein Effekt, der in einer Studie beobachtet wurde, auch ohne Beeinflussung von selbst aufgetreten wäre. In der medizinischen Forschung wird ein p-Wert, der unter 0,05, also unter 5% liegt, als Indiz für ein statistisch signifikantes Studienergebnis angesehen.
Noch wichtiger ist das 95%-Konfidenzintervall. Es beschreibt den Bereich von Werten, in dem man mit 95%iger Wahrscheinlichkeit sicher sein kann, dass darin das wahre Ergebnis für die gesamte Patientenpopulation liegt, aus der die Patienten für die Studien ausgewählt wurden.
Studienendpunkte
Studienendpunkte müssen sehr genau betrachtet werden. Sind es Surrogatparameter wie z.B. der Blutdruck oder Endpunkte wie z.B. die Lebensdauer? Endpunkte (Outcomes) sind die durch medizinische Maßnahmen erzielten Veränderungen des Gesundheitszustandes eines Patienten (wie z.B. Morbidität oder Mortalität), Surrogatparameter sind Zielgrößen, mit deren Hilfe eine Assoziation zu einem gewünschten Ereignis (meist Reduktion der Mortalität oder Morbidität) hergestellt wird. Ob eine Veränderung in einem Surrogatparameter dann auch tatsächlich relevante Folgen hat, ist damit noch nicht bewiesen. Studienergebnisse, die mithilfe von Surrogatparametern erhoben wurden, haben keine bedeutende klinische Relevanz.
Manipulation der Studienergebnisse
Um Manipulationen bei Studienergebnisse zu bemerken, müssen diese sehr genau gelesen werden. Als Beispiel führte Dietrich die Class-Studie auf. In dieser, auf 15 Monate angelegten Studie wurden Celecoxib, Ibuprofen und Diclofenac in Bezug auf ihre Nebenwirkungen miteinander verglichen. In den ersten Monaten sprachen die Studienergebnisse zu Gunsten von Celecoxib, am Studienende nicht mehr. Wird nun das Studienende "vorverlegt" (wie es geschehen war), werden andere Ergebnisse gefunden als nach dem regulären Ende.
Analogpräparate – Fortschritt oder Scheininnovationen?
Um Analogpräparate wird in Deutschland eine lebhafte gesundheitspolitische Diskussion geführt. Ermöglichen Analogpräparate eine bessere Therapie oder erhöhen sie lediglich die Arzneimittelkosten? Oder anders gefragt, nutzen Analogpräparate dem Hersteller oder dem Patienten? Diese Frage wird häufig emotional diskutiert, da teilweise die wissenschaftliche Ausgangsbasis für eine rationale Diskussion fehlt. Dr. Holger Gothe, Berlin, stellte eine Studie zur Marktbewertung von Analoga vor, in der die medizinische und wirtschaftliche Bedeutung von Analogpräparaten untersucht wurde.
In dieser Studie soll das Verhalten eines Arzneimittels auf dem Markt näher untersucht werden. Etwas vereinfacht lautet die Frage: Wie reagiert der Markt auf Angebot und Nachfrage, wenn Original- und Analogpräparate zur Verfügung stehen? Die Studie wurde vom Institut für Gesundheits- und Sozialforschung GmbH, Berlin (IGES) durchgeführt. Für die Untersuchung wurden für sämtliche seit 1980 auf den Markt gekommenen Fertigarzneimittel die Zahlen über abgesetzte Mengen und Preise ermittelt. Alle im Beobachtungszeitraum in den Markt eingeführten Wirkstoffe wurden nach der ATC-Klassifikation in Wirkstoffgruppen neu eingeteilt, um Vergleichsmöglichkeiten zu schaffen.
Zwischen 1980 und 2000 waren in Deutschland 453 Wirkstoffe neu auf den Markt gekommen. Davon haben 70 eine neue Wirkstoffgruppe begründet. Für 16 dieser 70 Wirkstoffgruppen kamen im betrachteten Zeitraum Generika auf den Markt. Die weitere Datenanalyse beschränkt sich auf diese 16 Wirkstoffgruppen, das heißt also auf diejenigen Wirkstoffgruppen, bei denen Originalpräparate und Generika im Handel sind. In 11 dieser Gruppen sind Analogpräparate (insgesamt 50) auf den Markt gekommen, in den anderen 5 Gruppen nicht.
Unterschiedliches Marktverhalten
Anhand einiger Wirkstoffgruppen erläuterte Gothe das unterschiedliche Marktverhalten und wie sich dieses auf Umsatzmengen sowie Preisgestaltung von Originalpräparat, Generikum und Analogpräparat auswirkt. Beispiel SSRI: Das Originalpräparat wurde bereits kurze Zeit nach seiner Einführung aus dem Handel genommen, erst durch die Einführung von Analogpräparaten wurden vermehrt SSRI verordnet; Analogwirkstoffe bestimmen heute den Markt. Beispiel H2-Blocker: Die Verordnungen des Originalpräparats Tagamet® nehmen ab, diejenigen der Generika mit Cimetidin zu, Analoga spielen keine Rolle. Beispiel Protonenpumpenhemmer: Der Wettbewerb zwischen Original- und Analogwirkstoffen in der Zeit vor der Einführung der Generika führte zu Preissenkungen und zu einer vermehrten Verordnung preisgünstiger Analogpräparate.
Einsparmöglichkeiten
Das Errechnen von Einsparmöglichkeiten durch Analoga ist erst ab 1989 sinnvoll, als eine stärkere staatliche Regulierung des GKV-Arzneimittelmarktes begonnen hatte. Hier führte Gothe wiederum einige Beispiele auf. Beispiel Protonenpumpenhemmer: Als Folge des Wettbewerbs, der durch Analogwirkstoffe ausgelöst wurde, konnten die Krankenkassen insgesamt ca. 400 Mio. DM einsparen. Beispiel Benzodiazepinrezeptor-Agonisten: Hier führte die Einführung von Analogwirkstoffen zu Mehrausgaben von rund 60 Mio. DM. Diese zwei Beispiele zeigen, dass Analogpräparate sowohl zu Einsparungen als auch zu Mehrausgaben führen können.
Analoga differenziert betrachten
Analogwirkstoffe werden differenziert verordnet. Es gibt Wirkstoffgruppen wie ACE-Hemmer oder H2-Blocker, in denen Analogwirkstoffe gegenüber dem Original und seinem Generikum nur einen geringen Marktanteil erreichen. In anderen Wirkstoffgruppen dagegen wie zum Beispiel bei den SSRI besetzten die späteren Analoga den Markt (Abb. 1). Dasselbe gilt für Antihistaminika, bei denen überwiegend Analogpräparate und/oder Generika verordnet werden. Gothe wertet dies als Beweis, dass Analoga sehr differenziert betrachtet und sinnvoll eingesetzt werden.
(H2B: H2-Blocker; PPI: Protonenpumpenhemmer; ACE-I: ACE-Hemmer;
SSRI: Selektive Serotonin-Wiederaufnahmehemmer; BRA: Benzodiazepinrezeptor-
Agonisten; AHI: Antihistaminika)
Pharmazeutische Bewertung
Die Sicherung und Aufrechterhaltung der pharmazeutischen Qualität gehört zu den täglichen Aufgaben des Apothekers. Prof. Dr. Manfred Schubert-Zsilavecz vom Zentrallaboratorium Deutscher Apotheker in Eschborn (ZL) stellte einige Bereiche vor, auf die ein besonderes Augenmerk zu richten ist.
Schätzungsweise gelangen pro Jahr 500 Tonnen Drogen der traditionellen chinesischen Medizin (TCM) nach Deutschland. Bei vielen Drogen kann die genaue Herkunft nicht mehr festgestellt werden; teilweise findet man hohe Belastungen mit Schwermetallen und Pestiziden. Rund 100 der traditionell eingesetzten chinesischen Drogen sind potenziell toxisch, als Beispiel nannte Schubert-Zsilavecz unter anderem Daphnis genkwae flos (enthält kanzerogene Diterpenester), Asari herba (mit den bedenklichen Inhaltsstoffen Safrol und verwandten Alkylenbenzolen), Pflanzen der Gattungen Senecio, Crotalaria und Heliotropium, die Pyrrolizidinalkaloide enthalten.
Weitere Gefahren bestehen durch Verwechslungen oder falsche Rezepturzusammenstellungen. Eine Untersuchung des ZL zeigte, dass viele Prüfzertifikate nicht den Anforderungen der Apothekenbetriebsordnung entsprechen. Ferner wurden Kontaminationen mit Aristolochiasäuren, Cadmium, Quecksilber und Aflatoxinen nachgewiesen.
Traditionelle chinesische Medizin
- Die TCM verwendet über 6000 Arzneien aus überwiegend pflanzlichen, seltener tierischen und mineralischen Ausgangsstoffen.
- Die Rezepturen sind aus mehreren Einzelmitteln zusammengesetzt.
- Von den über 600 verwendeten Arzneipflanzen sind 100 potenziell toxisch.
- Rechtliche Situation: Die importierten unbehandelten Pflanzenteile sind keine Arzneimittel im Sinne des § 2 AMG. Aber: Eine Mischung aus unterschiedlichen Pflanzenteilen aufgrund einer ärztlichen Verordnung wird als Arzneimittel betrachtet.
Arzneimittel für Kinder
Das ZL untersuchte 56 Trockensäfte mit Amoxicillin; 35 davon waren Monopräparate, 21 mit Clavulansäure kombiniert. Diese Säfte wurden nach unterschiedlichen Kriterien geprüft:
- Sekundärverpackung,
- Dosierhilfe,
- Dosiergenauigkeit,
- Sedimentationsverfahren,
- In-use-Stabilität.
Die Sekundärverpackung war in den meisten Fällen nicht zu beanstanden; in einigen Fällen hatte die Dosierhilfe keine Graduierung, obwohl dies auf der Packung angegeben war. 40 Säfte hatten einen kindersicheren Verschluss, 15 nicht. Die Dosierungsgenauigkeit wurde einmal mithilfe einer kalibrierten Glaspipette und im Vergleich dazu mit der beigefügten Dosierhilfe ermittelt. Eine genaue Dosierung wurde nur bei der Messgröße 1 Löffel erzielt; bei Messgrößen wie 1/2 oder 1/4 Messlöffel wurden hohe Überdosierungen festgestellt.
Die Empfehlung von Schubert-Zsilavecz: Zum korrekten Abmessen der Antibiotikasäfte eine Spritze verwenden. Bei manchen angefertigten Trockensäften ist die Bildung eines Sediments nicht sichtbar; daher sollen Säfte direkt vor der Applikation immer geschüttelt werden. Clavulansäure ist extrem thermolabil. Wird der angefertigte Saft nicht im Kühlschrank aufbewahrt, tritt ein starker Wirkverlust ein, der den Therapieerfolg gefährden kann.
Tipps für die Praxis
- Zur Dosierung von Antibiotikasäften nicht die beigelegte Dosierhilfe, sondern eine Spritze verwenden; dies gilt vor allem beim Abmessen kleiner Mengen.
- Angefertigte Trockensäfte vor der Applikation immer schütteln.
- Auf die korrekte Lagerung hinweisen.
Reihenuntersuchungen
Das ZL führt regelmäßig Reihenuntersuchungen durch. Bei der Untersuchung von Fertigarzneimitteln werden unter anderem Gehalt, Freisetzung, Zerfall, Verunreinigungen und Sekundärverpackungen untersucht. Schubert-Zsilavecz stellte einige in jüngster Zeit durchgeführten Untersuchungen zu Fertigarzneimitteln mit Acetylsalicylsäure, Enalapril, Hydrochlorothiazid und Ciprofloxazin vor, bei denen durchgehend eine hohe pharmazeutische Qualität nachgewiesen wurde. Eine Untersuchung von 30 Ibuprofenpräparaten ergab bei zwei Fertigarzneimitteln einen abweichenden Gehalt, woraufhin die Charge zurückgerufen wurde.
Phytopharmaka müssen dieselbe pharmazeutische Qualität aufweisen wie chemisch-synthetische Arzneimittel. In einer Reihenuntersuchung wurde die Qualität von apothekenpflichtigen und freiverkäuflichen Johanniskrautpräparaten untersucht. Die apothekenpflichtigen Johanniskrautpräparate entsprachen der geforderten Qualität, die freiverkäuflichen nicht. Die freiverkäuflichen Präparate hatten meist einen zu geringen Wirkstoffgehalt und zeigten keine Chargenkonformität.
Rezepturringversuch
In einem bundesweit ersten Rezepturringversuch sollte eine cortisonhaltige Salbe nach NRF 11.76 (hydrophile Clobetasolpropionat-Creme 0,05%) hergestellt werden. An dem Versuch beteiligten sich über 1000 Apotheken. Die eingesandten Salben wurden unter anderem auf ihre Identität, ihren Gehalt und die korrekte Deklaration hin überprüft. Die endgültige Auswertung der Ergebnisse steht noch aus, Trends können bereits festgestellt werden: Die Deklaration war in den meisten Fällen fehlerfrei. Identität und Gehalt entsprachen in mehr als 80% den Arzneibuchanforderungen. Bei Gehaltsabweichungen wurden eher Überdosierungen als Unterdosierungen festgestellt.
Pharmakoökonomische Beurteilung
Mithilfe pharmakoökonomischer Studien sollen die Ressourcen im Gesundheitswesen sinnvoll gesteuert werden. Dipl.-Ökonom Christoph Vauth, Forschungsstelle für Gesundheitsökonomie und Gesundheitssystemforschung, Hannover, gab einen Überblick zu den wesentlichen Studientypen und stellte das Konzept des Health Technology Assessment vor, mit dem eine Art integrierte Beurteilung von Arzneimitteln nach medizinischen und ökonomischen Gesichtspunkten vorgenommen wird.
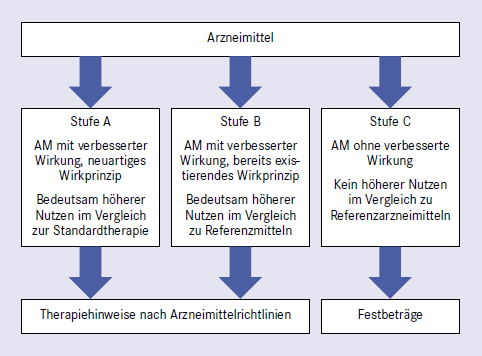
Ansteigende Kosten im Gesundheitswesen und Ausfälle bei der gesetzlichen Krankenversicherung stellen Politik und Akteure im Gesundheitswesen vor große Herausforderungen. Vor diesem Hintergrund sind diverse Maßnahmen wie zum Beispiel die Einführung von Disease Management Programmen (DMP), die Nutzenbewertung von Arzneimitteln nach § 35 SGB V (Abb. 2) und die Diskussion um die "vierte Hürde" zu sehen. In Anlehnung an die drei Hürden Qualität, Wirksamkeit und Unbedenklichkeit bei der Arzneimittelzulassung versteht man unter der "vierten Hürde" eine institutionalisierte Entscheidung über die Erstattungsfähigkeit eines Arzneimittels bei der beanspruchten Indikation.
GKV-Versorgung nach § 35b SGB V.
Health Technology Assessment (HTA)
HTA ist eine Methodik zur systematischen und transparenten Bewertung medizinischer Verfahren und Technologien, um Entscheidungsprozesse in diesem Bereich unter medizinischen, ökonomischen, juristischen, sozialen und ethischen Aspekten zu unterstützen. Der Begriff Technologie umfasst in diesem Kontext Medikamente, Instrumente, Prozeduren, Verfahren und Organisationssysteme. Es werden mehrere Zielgruppen angesprochen, als Beispiel führte Vauth Entscheidungsträger, Leistungsanbieter, Kostenträger, Mitarbeiter in medizinischen Heilberufen, Industrie sowie Patienten und Konsumenten auf. Ziele von HTA sind
- der Aufbau eines Informationssystems zur Bewertung medizinischer Verfahren und Technologien,
- das Erstellen von Informationen für Patienten und Konsumenten,
- die Unterstützung bei gesundheitspolitischen Entscheidungen und
- das Bereitstellen valider Informationen.
Mithilfe von HTA sollen in diesem Jahre diverse Fragestellungen bearbeitet werden.
Als Beispiele aus dem gesundheitspolitischen und medizinischen Bereich führte Vauth einige Arbeitsthemen auf:
- Verfahren zur Steigerung der Teilnahmerate an Krankheitsfrüherkennungsprogrammen
- Methoden der vergleichenden Bewertung pharmazeutischer Produkte
- Prävention rezidivierter Rückenschmerzen
- Kognitives Training bei Demenzen
- Misteltherapie zur Reduktion der Nebenwirkungen bei Krebserkrankungen
- Antioxidative Vitamine in der Prävention kardiovaskulärer Erkrankungen
- Neuraminidasehemmer in der Therapie und Postexpositionsprophylaxe der Influenza.
Leitlinien: Qualitätssicherung oder Listenmedizin?
Durch medizinische Leitlinien soll die Qualität in der Patientenversorgung verbessert und gesichert werden. Dies setzt aber bestimmte qualitative Anforderungen an die Leitlinie beziehungsweise an das Zustandekommen einer Leitlinie voraus.
Eine Hauptaufgabe des neu gegründeten Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen ist die Prüfung von Leitlinien. Wie dabei vorgegangen wird, erläuterte Dr. Hanna Kirchner vom IQWiG in Köln.
Zurzeit sind schätzungsweise über 1500 Leitlinien abrufbar. Leitlinien zu einem bestimmten Krankheitsbild empfehlen nicht immer das gleiche Vorgehen, was Kirchner am Beispiel der Otitis media im Kindesalter erläuterte. Die Leitlinien der deutschen HNO-Gesellschaft empfehlen die Gabe von Antibiotika, eine schottische Leitlinie lehnt sie ab, und bei den Vorgaben der amerikanischen Pädiater wird die Antibiotikatherapie als eine mögliche Option eingestuft. Die teilweise widersprüchlichen Angaben in Leitlinien und deren unterschiedliche Qualität sind verwirrend und führen zu Akzeptanzproblemen. Abhilfe kann hier das neu gegründete Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) schaffen.
Medizinische Leitlinien
- sind definiert als "systematisch entwickelte Entscheidungshilfen für Leistungserbringer und Patienten über eine angemessene Vorgehensweise bei speziellen Gesundheitsproblemen"
- sind geeignet, um Informationen und wissenschaftliche Standards zu vermitteln
- sollen die Qualität der Versorgung verbessern
- unterstreichen die Stellung des Patienten
- fördern eine qualitativ hoch stehende Arbeitsweise (GMP)
Das IQWiG
Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) prüft Leitlinien, bevor sie als Grundlage für gesundheitspolitische Entscheidungen herangezogen werden. Die gesetzliche Grundlage zur Schaffung dieses Instituts ist in § 139a SGB V verankert. Das IQWiG soll eine Qualitäts- und Nutzenbewertung vornehmen. Die Entscheidung, ob eine Leitlinie angenommen wird oder nicht, liegt beim Gemeinsamen Bundesausschuss (G-BA).
Ferner soll das IQWiG eine Nutzenbewertung von Arzneimitteln zu speziellen therapeutisch-pharmakologischen Fragen vornehmen. Es arbeitet wissenschaftlich unabhängig und soll zu mehr Transparenz und Patientenorientierung im Gesundheitswesen beitragen.
Über die Webseite www.iqwig.de können alle vom Institut bearbeiteten Publikationen zu Arzneimitteln, Leitlinien, diagnostischen und therapeutischen Maßnahmen abgerufen werden.
Herausgeber medizinischer Leitlinien
- Medizinisch-wissenschaftliche Fachgesellschaften (AMWF)
- Ärztliche Berufsverbände
- Berufsgenossenschaften
- Rentenversicherer
- Krankenhäuser
- Arztnetze
- Qualitätszirkel
- Nationales Programm für Versorgungsleitlinien
- Arzneimittelkommission der deutschen Ärzteschaft
Wie entstehen Leitlinien?
Für eine bestimmte Fragestellung oder für ein bestimmtes Krankheitsbild werden alle wissenschaftlichen Informationen zusammengetragen, bewertet und nach methodischen Gesichtspunkten zusammengefasst. Dann wird die Evidenzgrundlage der Informationen überprüft. Dem folgt die Ausarbeitung der Leitlinie durch Vertreter aller in Frage kommenden Fachdisziplinen und in Zusammenarbeit mit Patienten. Die ausgearbeiteten Leitlinien müssen gewissen Qualitätsanforderungen entsprechen, umsetzbar sein und regelmäßig überprüft werden. Die Autoren von Leitlinien sollten unabhängig sein, Verflechtungen oder Abhängigkeiten müssen dargelegt werden. Die ausgearbeitete Leitlinie geht nun an den Gemeinsamen Bundesausschuss (G-BA), bei dem die Entscheidung über die Umsetzung liegt.
Leitlinien zur Kostensenkung?
Die Auswirkungen einer Medizin nach Leitlinien werden unterschiedlich eingeschätzt. Gegner dieses Vorgehens sehen in der "Listenmedizin" ein politisches Instrument zur Einschränkung der Therapiefreiheit und bezweifeln, ob eine Therapie nach Leitlinien zu einer Senkung der Kosten im Gesundheitswesen führt. Die Befürworter der Leitlinien sehen in diesen ein Mittel zur Qualitätssicherung und zu einer transparenten, evidenzbasierten Medizin.
Ob durch die Anwendung der Leitlinien Kosten gespart werden, ist fraglich. So führte Dr. Dietrich eine Studie aus Mecklenburg-Vorpommern an, in der exemplarisch für ein Krankheitsbild aufgezeigt wurde, dass eine Therapie nach Leitlinien zu einem deutlichen Kostenanstieg führt. Dasselbe dürfte der Fall sein, wenn Osteoporosepatienten einer Leitlinie folgend vermehrt mit teuren, aber effektiven Bisphosphonaten behandelt werden.
Internet Webseiten und Datenbanken
Arzneimittelkommission der deutschen Ärzteschaft www.akdae.de
- Cochrane Library www.cochrane.org
- SIGN (Scottish Intercollegiate Guidelines Network) www.sign.ac.uk
- NICE (National Institute for Clinical Excellence) www.nice.org.uk
- CRD (Centre for Reviews and Dissemination) www.york.ac.uk/inst/crd
Internet
- Den größten Überblick zu bestehenden Leitlinien bietet die Website des internationalen Leitliniennetzwerks GIN (Guidelines International Network): www.g-i-n.net. Über die Guideline Library haben Mitglieder des Netzwerks freien Zugang zu über 2400 Leitlinien aus 29 Ländern.
- Ärztliches Zentrum für Qualität in der Medizin (ÄZQ) in Berlin; das deutschsprachige Portal ist über www.leitlinien.de zu erreichen und bietet über Links den Zugang zu allen bekannten deutschen und internationalen Leitlinien.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.