













- DAZ.online
- DAZ / AZ
- DAZ 18/2014
- Lesezeichen im Genom
UniDAZ
Lesezeichen im Genom
Struktur-Wirkungs-Beziehungen an Hemmstoffen von Histon-Demethylasen und Histon-Desacetylasen
Editorial
Liebe Leserinnen, liebe Leser!
Der Siegerbeitrag des diesjährigen UniDAZ-Wissenschaftspreises ging aus einer Bachelorarbeit hervor, die Eva-Maria Herrlinger am Lehrstuhl für Pharmazeutische und Medizinische Chemie der Albert-Ludwigs-Universität Freiburg unter der fachlichen Leitung von Herrn Prof. Dr. Manfred Jung verfasst hat.
Der UniDAZ-Wissenschaftspreis wurde ihr auf der Interpharm in Berlin verliehen (siehe DAZ 2014, Nr. 14, S. 78).
Herrlinger untersuchte die Struktur-Wirkungs-Beziehungen an Hemmstoffen von Histon-Demethylasen und Histon-Desacetylasen. Diese Enzyme katalysieren die Abspaltung modifizierender Gruppen der Histone. Hierdurch wird es möglich, dass ein Gen abgelesen werden soll. Damit spielen diese Enzyme eine wichtige Rolle bei der Steuerung von Zellfunktionen. Um ihre genauen Funktionen aufzuklären, ist die Suche nach selektiven Hemmstoffen erforderlich – ein wichtiger Forschungsschwerpunkt der Epigenetik. Herrlinger modifizierte einen kürzlich beschriebenen Histondesacetylase-Hemmer und untersuchte die Hemmwirkungen von zwei neu synthetisierten Derivaten. In diesem Beitrag erfahren Sie, welche molekularen Strukturen die Wirksamkeit der Hemmstoffe beeinflussen.
Lesen Sie, wie die Hemmwirkungen zustande kommen und wie man sie messen kann.
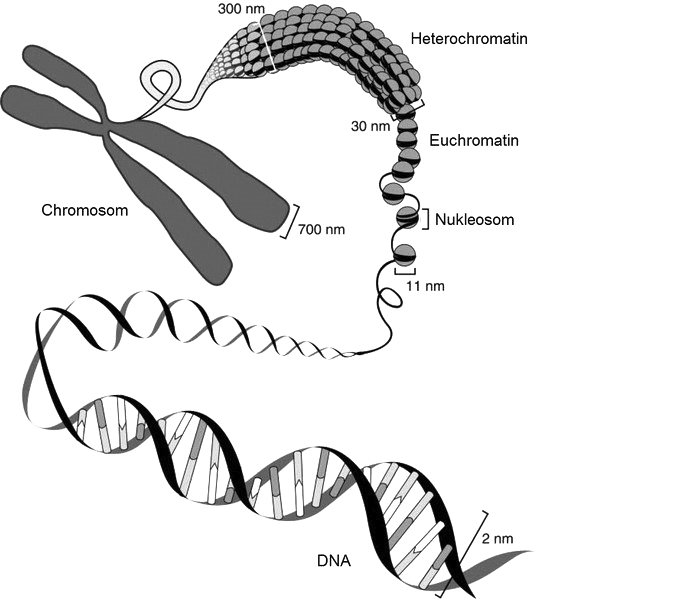
Die Desoxyribonukleinsäure (DNA) ist in Eukaryonten mit Proteinen zu einer kompakten Struktur, dem Chromatin, verbunden (s. Abb. 1). Die Basiseinheit des Chromatins stellen die Nukleosome dar, die durch Linker-DNA miteinander verbunden sind. Ein Nukleosom besteht aus einem Histon-Oktamer und der sich darum herumwindenden DNA. Es gibt zwei Arten von Chromatin:
- das stärker kondensierte Heterochromatin, das die Transkription der Gene behindert, sowie
- das Euchromatin, dessen Struktur lockerer ist, was die Transkription begünstigt [1].
Wie stark das Chromatin kondensiert ist, hängt – neben der DNA-Methylierung – vor allem von den N-terminalen Enden der Histone ab, die mit benachbarten Nukleosomen interagieren [2]. Die N-terminalen Enden ragen aus dem Komplex heraus und können posttranslational modifiziert werden [2].
Quelle: Modifiziert nach [3]
Modifikation durch Methylierung und Acetylierung
Die am häufigsten vorkommenden Modifikationen am N-terminalen Ende von Histonen sind die Methylierung und Acetylierung der Aminosäure Lysin.
Die Acetylierung wird durch Histon-Acetyltransferasen katalysiert und bewirkt eine Neutralisation der positiven Ladung am Stickstoff [5]. Dadurch nimmt die Affinität des Histonendes zu benachbarten Nukleosomen und zur negativ geladenen DNA ab, sodass die Chromatinstruktur lockerer wird [6]. Die Acetylierung spielt daher eine wichtige Rolle bei der Wandlung vom Heterochromatin zum Euchromatin und ermöglicht die Bindung von regulatorischen Proteinen an die DNA und somit die Genaktivierung [5].
Die Abspaltung der Acetylgruppe – also der umgekehrte Vorgang – wird durch Histon-Desacetylasen (HDAC) katalysiert.
Die Methylierung am ε-Stickstoff der Aminosäure Lysin wird durch Methyltransferasen katalysiert [7]. Die Methylierungsmuster fungieren als Bindestelle für andere regulatorische Proteine, die die Struktur des Chromatins beeinflussen und die Transkription steuern [8].
Die Abspaltung der Methylgruppen wird durch Demethylasen katalysiert, zu denen auch die Jumonji-C-Domäne-enthaltenden Demethylasen (JmjD) gehören. Bei den JmjD handelt es sich um Oxygenasen, die ein katalytisches Fe2+ im aktiven Zentrum – der JmjC-Domäne – besitzen und α-Ketoglutarat (α-KG) als Kosubstrat verwenden [5, 8].
Individuelle Genexpressionsmuster
Alle Zellen im menschlichen Körper besitzen das gleiche Erbgut in Form von DNA. Trotzdem unterscheiden sie sich in ihrer biologischen Funktion, die sie im Gewebe und den Organen einnehmen, ohne dass die DNA-Sequenz verändert wird [5]. Die Unterschiede sind auf die individuellen Genexpressionsmuster der verschiedenen Zellen zurückzuführen, die durch die reversible DNA-Methylierung und den Kondensationsgrad des Chromatins beeinflusst werden [4, 5]. Folglich spielt die epigenetische Regulation eine wichtige Rolle während der zellulären Differenzierung und Entwicklung [4, 5]. Außerdem steuert sie auch zelluläre Funktionen wie die Immunantwort und die Signaltransduktion [4]. Bei einer Fehlregulation kann es zu verschiedenen Krankheiten kommen, die von neurodegenerativen Erkrankungen bis hin zu Krebs reichen [5, 9]. Ein pathologisch verändertes Genexpressionsmuster wird entweder durch eine Mutation der Regulator-Proteine oder durch deren veränderte Expression oder durch deren erhöhte Aktivität – aufgrund von Transkriptionsfaktoren oder äußeren Reizen – verursacht [5].
Hemmstoffe von epigenetischen Enzymen
Ein spezielles Forschungsgebiet sind Hemmstoffe von epigenetischen Enzymen (Regulator-Proteinen), die an dem Krankheits-assoziierten epigenetischen Zustand der Zelle beteiligt sind [5].
Die ersten JmjD-Inhibitoren wurden 2008 publiziert und basieren auf Substanzen, die bekannte Inhibitoren anderer Eisen(II)- und α-KG-abhängiger Enzyme darstellen [12, 13]. Da die meisten Inhibitoren Substrat- oder Kosubstrat-Imitatoren sind, die das Enzym kompetitiv hemmen, zeigen sie auch Aktivität gegenüber anderen Enzymfamilien, sodass sie nur eine geringe Spezifität aufweisen. Durch die Entwicklung Subtypen-selektiver Hemmstoffe könnte die Funktion der JmjD besser untersucht werden, und möglicherweise könnten dabei auch potenzielle Pharmaka gefunden werden.
Da viele HDAC ein Zn2+ im aktiven Zentrum besitzen, haben die meisten HDAC-Inhibitoren eine Zink-bindende Gruppe [14]. Ein bereits 2006 von der amerikanischen Arzneimittelbehörde FDA zugelassener HDAC-Hemmstoff ist SAHA (suberoylanilide hydroxamic acid, INN: Vorinostat, Zolinza®), der gegen das kutane T-Zell-Lymphom eingesetzt wird [15]. SAHA kann das Tumorwachstum anhalten und die Apoptose der Tumorzellen induzieren. Daher ist die Suche nach weiteren selektiven HDAC-Hemmstoffen ein vielversprechendes Forschungsfeld der Onkologie [16]. HDAC-Hemmstoffe besitzen außerdem das Potenzial, neurologische Erkrankungen zu heilen und die Gedächtnisleistung zu steigern (cognitive enhancers) [17].
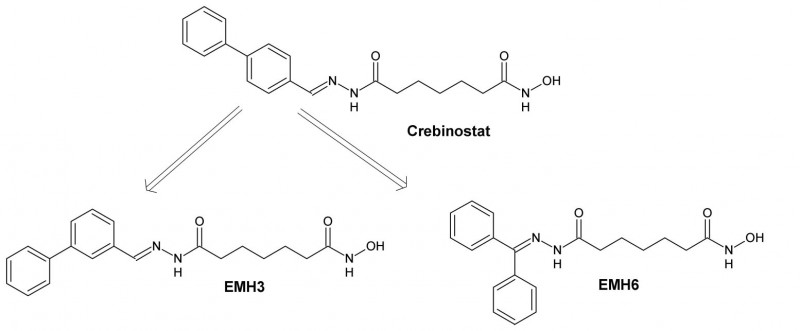
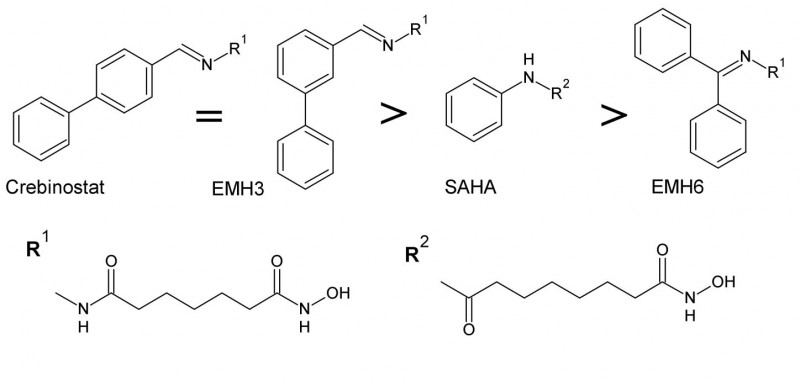
Variation des HDAC-Inhibitors Crebinostat
Crebinostat wurde letztes Jahr als HDAC-Inhibitor vorgestellt [19], nachdem D. Stolfa und M. Roatsch ihn bereits als potenziellen Hemmstoff von JmjD2A erkannt hatten. Auch andere HDAC-Inhibitoren, die wie Crebinostat eine Hydroxamsäure als Zink-chelatisierende Gruppe haben, wurden als potenzielle JmjD-Hemmstoffe beschrieben [12, 13]. Diese Wirkung ist wahrscheinlich darauf zurückzuführen, dass sie das in den JmjD enthaltene Eisen chelatisieren [12].
Quelle: Herrlinger
Ausgehend von Crebinostat, das am Hydrazon ein Biphenyl besitzt, sollte durch Variation der aromatischen Seitenkette untersucht werden, welchen Einfluss diese auf die Aktivität und Selektivität des Hemmstoffes hat (Struktur-Wirkungs-Beziehung). Deshalb wurden die Crebinostat-Derivate EMH3 und EMH6 synthetisiert (s. Abb. 2). Das C7-Grundgerüst der Hydroxamsäure blieb dabei unverändert.
Die Hemmwirkungen der Crebinostat-Derivate wurden in vitro getestet. Dabei wurde ihre Selektivität zwischen JmjD2A, HDAC1 und HDAC6 verglichen.
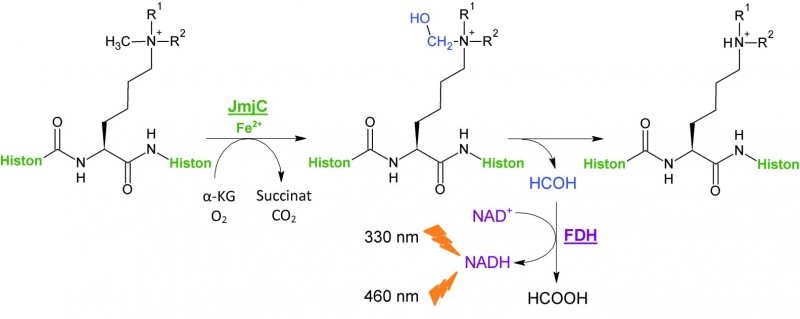
JMJ-FDH-Assay
Beim JMJ-FDH-Assay handelt es sich um einen enzymgekoppelten Assay, der auf bereits publizierten Methoden basiert [10, 12, 21]. Bei jeder Demethylierung des Substrates durch JmjD entsteht ein Molekül Formaldehyd [8], das durch eine Formaldehyd-Dehydrogenase (FDH)-gekoppelte Reaktion detektiert werden kann (s. Abb. 3): Während die FDH das entstandene Formaldehyd oxidiert, wird der FDH-Kofaktor Nicotinamid-Adenin-Dinukleotid (NAD+) zum fluoreszierenden NADH reduziert [22]. Je aktiver die JmjD2A ist, desto mehr Formaldehyd entsteht, und desto höher ist die gemessene Fluoreszenz.
Durch Messung einer Verdünnungsreihe des Hemmstoffs kann eine IC50-Kurve erstellt werden. Der IC50-Wert entspricht der Konzentration des Hemmstoffes (inhibitory concentration), bei der das Enzym nur noch 50% seiner eigentlichen Aktivität besitzt.
Quelle: Modifiziert nach [21|
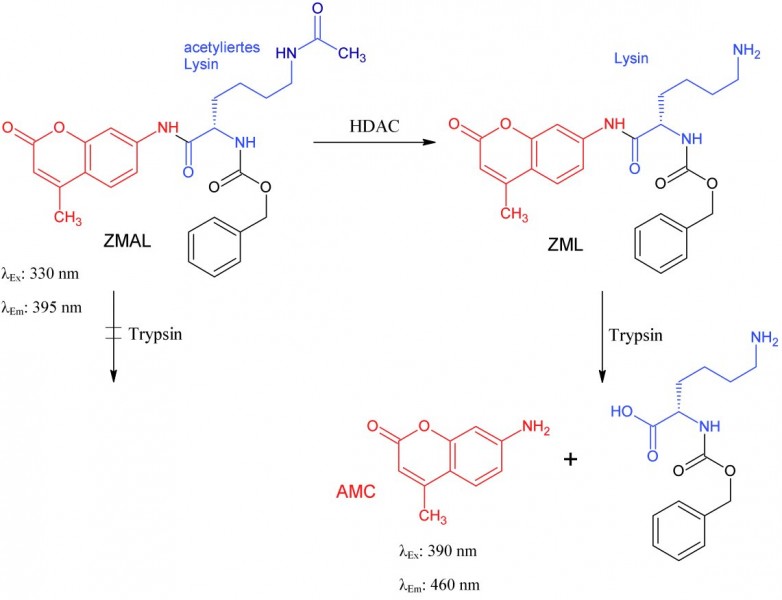
Trypsin-Assay
Beim Trypsin-Assay (Schema in Abb. 4) handelt es sich um eine Variante des histone deacetylase assay – homogeneous (HDASH) [23], der im Arbeitskreis M. Jung in Freiburg entwickelt wurde [24]. Das Prinzip des Assays besteht darin, die umgesetzte Menge an Substrat zu messen, um auf die Aktivität der HDAC zu schließen. Als Substrat wird ZMAL verwendet, ein acetyliertes Derivat der Aminosäure Lysin, welches den Fluorophor 7-Amino-4-methylcumarin (AMC) enthält. Um die Menge des desacetylierten Metaboliten von ZMAL, des ZML, zu detektieren, wird die Endopeptidase Trypsin verwendet [25]. Trypsin erkennt und spaltet ZML (nicht aber ZMAL), wobei AMC frei wird [24]. Durch Fluoreszenzmessung kann die AMC-Konzentration und damit die HDAC-Aktivität bestimmt werden [24].
Quelle: Modifiziert nach [24|
Ergebnisse der Struktur-Wirkungs-Beziehungen
Die Auswertung der Versuche mit dem JMJ-FDH-Assay und dem Trypsin-Assay erfolgte mit dem Programm GraphPad Prism 4. Die gemessenen IC50-Werte sind in der Tabelle zusammengefasst; zudem wurden die Substanzen gemäß ihrer Hemmwirkung sowohl auf die beiden HDAC als auch auf JmjD2A in einer Reihenfolge sortiert (s. Abb. 5).
Quelle: Herrlinger
EMH3 zeigte, verglichen mit EMH6 und SAHA, stärkere Hemmwirkungen sowohl auf HDAC als auch auf JmjD2A. Ein direkter Vergleich der IC50-Werte bei HDAC1 und HDAC6 ist hier nicht möglich, da die gemessenen Enzymaktivitäten in der DMSO-Kontrolle (Dimethylsulfoxid) nicht identisch waren.
Beide synthetisierten Hemmstoffe, EMH3 und EMH6, sind bessere HDAC-Inhibitoren als JmjD2A-Inhibitoren, was aufgrund der Hydroxamsäure-Funktion auch zu vermuten war.
Zwischen der meta-Substitution (bei EMH3) und der para-Substitution des Biphenyls (bei Crebinostat) war kein Unterschied bezüglich der Hemmwirkung auf JmjD2A erkennbar.
Für HDAC1 und HDAC6 ist eine entsprechende Feststellung nicht möglich, da keine Messwerte für Crebinostat mit dem gleichen Assay vorliegen (s. Tab.). In einem anderen In-vitro-Assay wurde bei Crebinostat eine sechsfach höhere HDAC-Hemmwirkung gemessen als bei SAHA [19].
Inhibitoren mit einem Biphenyl als Seitenkette sind wirksamer als Inhibitoren mit einem Phenyl (wie SAHA). Dies gilt für beide Enzymfamilien und konnte vor allem für die JmjD schon mehrfach belegt werden. Außerdem konnte gezeigt werden, dass ein aromatischer Rest am Hydrazon effektiver ist als zwei aromatische Reste (wie bei EMH6).
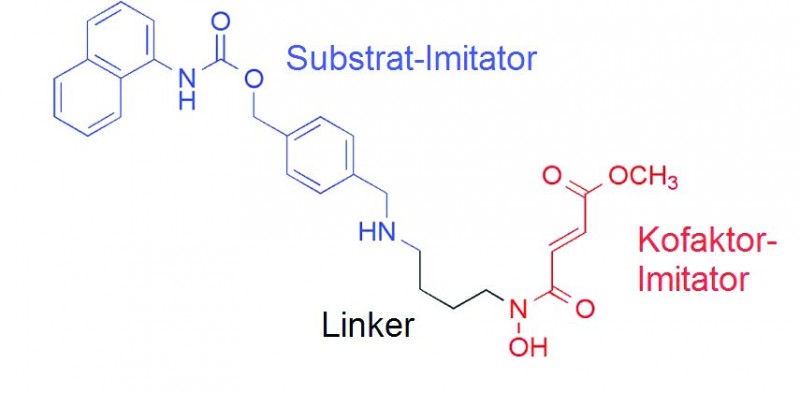
Erhöhung der JmjD-Selektivität
Um die Selektivität eines JmjD-Inhibitors zu erhöhen, bietet sich die Modifizierung seiner Hydroxamsäure an. Ein gutes Beispiel hierfür ist Methylstat [26] (s. Abb. 6), welches in vivo selektiv JmjD hemmt. Es wurde durch strukturbasiertes Design entwickelt, wobei der enzymatische Mechanismus und die Kristallstrukturen zugrunde gelegt wurden. Methylstat setzt sich aus einem Methyllysin-Imitator (Substrat-Imitator), einem α-KG-Imitator (Kofaktor-Imitator) und einem Verbindungsstück (Linker) zusammen, wobei der Linker die Geometrie und den relativen Abstand zwischen α-KG und Substrat in der Kristallstruktur imitieren soll. So kann die Selektivität eines JmjD-Inhibitors auch innerhalb der JmjD-Unterfamilien erhöht werden [26].
Quelle: Modifiziert nach [26]
Erhöhung der HDAC-Selektivität
Um die HDAC-Selektivität zu erhöhen, ist die Modifikation der Zink-bindenden Domäne ebenfalls eine Möglichkeit, da neben dem aktiven Zentrum des Enzyms eine hydrophobe „Tasche“ liegt, in der eine zusätzliche Seitenkette binden kann [14]. Da das aktive Zentrum und der hinführende Tunnel der HDAC hoch konserviert sind [27] und die Unterschiede eher in peripheren Regionen zu finden sind [18], könnten selektive Inhibitoren auch aus einer Sammlung von Stoffen identifiziert werden, deren Kopfgruppe – ähnlich wie in dieser Arbeit – variiert wurde [20].
Länge des Linkers und Substituent entscheidend
Zusammenfassend können zwei wichtige Eigenschaften eines guten Hemmstoffes für HDAC und JmjD2A festgehalten werden:
- Zum einen die richtige Länge des Linkers, der optimal in den hydrophoben Tunnel zum aktiven Zentrum passen sollte.
- Zum anderen die Substituenten, die mit dem aktiven Zentrum und Subtypen-spezifischen Enzymtaschen interagieren und dadurch selektiv das Enzym hemmen sollten.
Für ein erfolgreiches Hemmstoff-Design ist es somit wichtig, einen Einblick in die Strukturen der Enzyme zu erhalten, um auch elektronische und sterische Aspekte beachten zu können. Es wäre durchaus sinnvoll, virtuelle oder High-Throughput-Screenings durchzuführen, um interessante Substituenten zu identifizieren.
Quellen:
[1] Jenuwein T, Allis CD. Translating the Histone Code. Science 2001;293: 1074–1080.
[2] Hansen JC, Tse C, Wolffe AP. Structure and Function of the Core Histone N-Termini: More Than Meets the Eye. Biochemistry (Mosc.) 1998; 37: 17637–17641.
[3] Weier HUG. DNA Fiber Mapping Techniques for the Assembly of High-resolution Physical Maps. J. Histochem. Cytochem. 2001; 49: 939–948.
[4] Itoh Y, Suzuki T, Miyata N. Small-molecular modulators of cancer-associated epigenetic mechanisms. Mol. Biosyst. 2013; 9: 873-896.
[5] Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 2012; 11: 384–400.
[6] Hansen JC. Linking genome structure and function through specific histone acetylation. Acs Chem. Biol. 2006; 1: 69–72.
[7] Trievel RC. Structure and function of histone methyltransferases. Crit. Rev. Eukaryot. Gene Expr. 2004; 14: 147–169.
[8] Hoffmann I, Roatsch M, Schmitt ML, Carlino L, Pippel M, Sippl W, Jung M. The role of histone demethylases in cancer therapy. Mol. Oncol. 2012; 6: 683–703.
[9] Portela A, Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010; 28: 1057–1068.
[10] Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat. Struct. Mol. Biol. 2007; 14: 689–695.
[11] Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G. Structural Insights into Histone Demethylation by JMJD2 Family Members. Cell 2006; 125: 691–702.
[12] Rose NR, Ng SS, Mecinović J, Liénard BMR, Bello SH, Sun Z, McDonough MA, Oppermann U, Schofield CJ. Inhibitor Scaffolds for 2-Oxoglutarate-Dependent Histone Lysine Demethylases. J. Med. Chem. 2008; 51: 7053–7056.
[13] Schofield C, McDonough M, Rose N, Thalhammer A. Histone Lysine Demethylase Inhibitors. WO/2010/043866, April 22, 2010.
[14] Pontiki E, Hadjipavlou-Litina D. Histone deacetylase inhibitors (HDACIs). Structure - activity relationships: history and new QSAR perspectives. Med. Res. Rev. 2012; 32: 1–165.
[15] Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010; 6: 238–243.
[16] Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000; 60: 5165–5170.
[17] Gräff J, Tsai LH. The Potential of HDAC Inhibitors as Cognitive Enhancers. Annu. Rev. Pharmacol. Toxicol. 2013; 53: 311–330.
[18] Sternson SM, Wong JC, Grozinger CM, Schreiber SL. Synthesis of 7200 Small Molecules Based on a Substructural Analysis of the Histone Deacetylase Inhibitors Trichostatin and Trapoxin. Org. Lett. 2001; 3: 4239–4242.
[19] Fass DM, Reis SA, Ghosh B, Hennig KM, Joseph NF, Zhao WN, Nieland TJF, Guan JS, Groves Kuhnle CE, Tang W, Barker DD, Mazitschek R, Schreiber SL, Tsai LH, Haggarty SJ. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 2013; 64: 81–96.
[20] Tang W, Luo T, Greenberg EF, Bradner JE, Schreiber SL. Discovery of histone deacetylase 8 selective inhibitors. Bioorg. Med. Chem. Lett. 2011; 21: 2601–2605.
[21] Sakurai M, Rose NR, Schultz L, Quinn AM, Jadhav A, Ng SS, Oppermann U, Schofield CJ, Simeonov A. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol. Biosyst. 2010; 6: 357–364.
[22] Lizcano JM, Unzeta M, Tipton KF. A Spectrophotometric Method for Determining the Oxidative Deamination of Methylamine by the Amine Oxidases. Anal. Biochem. 2000; 286: 75–79.
[23] Heltweg B, Jung M. A Homogeneous Nonisotopic Histone Deacetylase Activity Assay. J. Biomol. Screen. 2003; 8: 89–95.
[24] Hauser AT, Gajer JM, Jung M. Nonradioactive In Vitro Assays for Histone Deacetylases. In Protein Acetylation; Methods in Molecular Biology; Humana Press, 2013; pp. 211–227.
[25] Heltweg B, Trapp J, Jung M. In vitro assays for the determination of histone deacetylase activity. Methods 2005; 36: 332–337.
[26] Luo X, Liu Y, Kubicek S, Myllyharju J, Tumber A, Ng S, Che KH, Podoll J, Heightman TD, Oppermann U, Schreiber SL, Wang X. A Selective Inhibitor and Probe of the Cellular Functions of Jumonji C Domain-Containing Histone Demethylases. J. Am. Chem. Soc. 2011; 133: 9451–9456.
[27] Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, Francesco RD, Gallinari P, Steinkühler C, Marco SD. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004; 101: 15064–15069.
Autorin
Eva-Maria Herrlinger studiert in Freiburg Pharmazeutische Wissenschaften und ist im zweiten Semester des Masterstudiums.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.