

















- DAZ.online
- DAZ / AZ
- DAZ 38/2015
- Hilfe bei seltenen ...
Orphan Drugs
Hilfe bei seltenen Krankheiten
Ein Einblick in die Entwicklung und Zulassung von Orphan Drugs

Foto: Štěpán Kápl – Fotolia.com
Disclaimer
Die im Folgenden geäußerten Sachverhalte sind persönliche Ansichten der Verfasser. Es handelt sich nicht um Ansichten des Bundesinstituts für Arzneimittel und Medizinprodukte, der European Medicines Agency oder eines ihrer Gremien oder Ausschüsse. Ein Rechtsanspruch entsteht nicht.
Arzneimittel gegen seltene Erkrankungen
Der englische Ausdruck „Orphan“ steht für „Waise“ oder „Waisenkind“. In der Arzneimitteltherapie wird dieser Begriff im Zusammenhang mit einer seltenen Erkrankung genutzt. Orphan Drugs sind Arzneimittel zur Behandlung von seltenen Erkrankungen (Orphan Diseases) und beziehen sich auf eine kleine Zielpopulation oder ein begrenztes Indikationsgebiet. Die Orphan Drugs sind so vielfältig wie die Indikationen selbst. Es wurden bereits Antikörper sowie Small Molecules zugelassen, es werden neue Arzneimittel entwickelt und „alte“ Arzneimittel finden neue Verwendung.
Zurzeit sind durch die Europäische Kommission 118 Orphan Drugs zugelassen und 1192 erhielten eine Ausweisung als Arzneimittel für seltene Erkrankungen (Orphan Designation [11]. Es ist anzumerken, dass es keine „Orphan-Medizinprodukte“ oder „Medizinprodukte gegen seltene Leiden“ gibt: die wörtliche Übersetzung von „Orphan Medicinal Products“ ins Deutsche ist irreführend. Medizinprodukte werden im englischen Medical Devices genannt. Lediglich Orphan Drugs können mit wirtschaftlichen Anreizen gefördert werden. Alle zugelassenen Arzneimittel für seltene Erkrankungen in der EU werden von der Europäischen Kommission im Community-Register geführt (http://ec.europa.eu/health/documents/community-register/html/orphreg.htm).
Obwohl Japan, Amerika und die EU-Länder Zulassungskriterien vereinheitlichen, bestehen Unterschiede der Beurteilungskriterien, da seltene Erkrankungen länderspezifisch definiert werden. Als „selten“ gilt eine Krankheit dann, wenn sie mit folgender Prävalenz auftritt:
- in der EU weniger als fünf/10.000 Einwohner,
- in den USA weniger als 7,5/10.000 Einwohner [4] und
- in Japan weniger als vier/10.000 Einwohnern.
Ebenfalls als „selten“ eingestuft wird eine Inzidenz von
- in der EU weniger als 230.000 Erkrankungen/Jahr,
- in den USA weniger als 200.000 Erkrankungen/Jahr [4] und
- in Japan weniger als 50.000 Erkrankungen/Jahr.
Regulatorische Historie der Orphan Drugs
Erstmals gesetzlich verankert wurden seltene Arzneimittel 1983 im Orphan Drug Act in den USA. Schon damals wurde festgestellt, dass nur wenige Pharmaunternehmen im Indikationsgebiet von seltenen Erkrankungen forschten und Arzneimittel entwickelten. Aufgrund der Erwartung von geringen Absatzzahlen wurde auf diesem Gebiet kaum investiert. Um den Erkrankten Therapiemöglichkeiten bieten zu können, wurden in den USA Gesetze geschaffen, um die fehlenden wirtschaftlichen Anreize zu kompensieren [4]. 1997 folgten Japan und Singapur diesem Beispiel. Nachdem 1998 Australien einen äquivalenten Gesetzesbeschluss verabschiedete, zog 1999 auch Europa nach.
2000 trat mit der EU-Verordnung EG/141/2000 die Orphan Regulation in Kraft (siehe Kasten „Orphan Regulation“). Die Verordnung ermöglicht wirtschaftliche Anreize, um finanzielle Nachteile des pharmazeutischen Unternehmens auszugleichen. Um diese Anreize nutzen zu können, muss ein Arzneimittel bestimmte Voraussetzungen erfüllen. Beispielsweise wurde in der EG/141/2000 auch definiert, dass eine seltene Erkrankung lebensbedrohlich oder zumindest schwerwiegend sein muss (keine „Bagatell-Erkrankungen“) [1].
Orphan Regulation
Orphan Diseases werden innerhalb der EU, Japans und der USA unterschiedlich definiert. Gemeinsam bilden diese Länder die „ICH-Region“ (ICH = International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). Ihr Ziel ist es, harmonisierte Kriterien für die Zulassung von Arzneimitteln zu erarbeiten. Zu diesem Zweck verfassen Experten der ICH-Region Guidelines zu diversen Themenkomplexen (pharmazeutische Qualität, Arzneimittelsicherheit, Wirksamkeit sowie interdisziplinäre Bereiche). Ende 1999 wurde vom Europäischen Parlament die sogenannte Orphan Regulation verabschiedet (EG/141/2000) [1]. Hierin wird zum einen festgelegt, welche Kriterien erfüllt werden müssen, damit ein Wirkstoff für seltene Erkrankungen verwendet werden kann. Kommt es zur Anerkennung eines solchen Status, spricht man von der Erteilung einer Orphan Designation. Zum anderen werden wirtschaftliche Anreize (Incentives) zur Entwicklung von Orphan Drugs definiert, Anreize für den pharmazeutischen Unternehmer trotz unwirtschaftlicher Ausgangssituation ein Entwicklungsprogramm für Arzneimittel gegen seltene Leiden aufzusetzen.
Vor der Beantragung einer Marktzulassung als Orphan Drug ist eine Ausweisung des Arzneimittels für eine seltene Erkrankung obligat (Orphan Designation). Dies ist keine Zulassungsart, sondern ein Antrag auf die Anerkennung, dass der entwickelte Wirkstoff ein Arzneimittel zur Behandlung einer seltenen Erkrankung ist (siehe Abbildung 1). Die Entscheidung fällt das Expertengremium Committee for Orphan Medicinal Products (COMP) bei der EMA. Es setzt sich aus je einem Vertreter der 28 Mitgliedsländer, drei Repräsentanten verschiedener Patientenorganisationen, drei von der EMA empfohlenen Mitgliedern und zwei Mitgliedern ohne Stimmrecht aus Norwegen und Island zusammen [3, 7].
Abb. 1: Zulassung eines Orphan Drug und die Voraussetzungen
Vorteile einer Orphan Designation
Die Orphan Designation eröffnet dem pharmazeutischen Unternehmer finanziellen Spielraum, denn die Vorteile umfassen z. B. vergünstigte Zulassungsgebühren und bevorzugten Zugang zu Förderprogrammen auf nationaler Ebene. Wie Tabelle 1 zeigt, gibt es nicht nur finanzielle Hilfen in Deutschland. Des Weiteren profitiert der pharmazeutische Unternehmer von gebührenfreiem Zugang zu wissenschaftlicher Beratung (Scientific Advice) durch die EMA. Diese Beratungsleistungen (Protocol Assistance) können die Planung klinischer Studien umfassen oder auch Kernpunkte der regulatorischen Route. Der wohl wirksamste Anreiz für einen pharmazeutischen Unternehmer ist die Marktexklusivität. Sie ist vom Patentschutz abzugrenzen, da es sich nicht um den Schutz einer chemischen Struktur, wie beim Patentschutz, sondern um den Schutz eines Indikationsgebietes handelt. Durch die zehnjährige Marktexklusivität wird der pharmazeutische Unternehmer gewissermaßen zum Monopolisten, das Arzneimittel wird gegen Me-too-Produkte geschützt. Diese bringen mit unwesentlichen Veränderungen in der Molekülstruktur und mit demselben Wirkungsmechanismus nur einen geringen (oder gar keinen) medizinischen Fortschritt. Die Aussicht auf Marktexklusivität wiederum ermöglicht oftmals leichteren Zugang zu Finanzierungsmöglichkeiten und steigert die Attraktivität für Investoren [2]. Seit der Einführung der frühen Nutzenbewertung nach dem Arzneimittelmarkt-Neuordnungsgesetz (AMNOG) gibt es einen weiteren Vorteil einer Orphan Designation. Da es per definitionem bei einem Arzneimittel gegen seltene Erkrankungen bisher keine oder nur unzureichende Therapiemöglichkeiten gibt, wird auf den Nachweis des „therapeutischen Zusatznutzens im Vergleich zur zweckmäßigen Vergleichstherapie“ verzichtet, solange der jährliche Umsatz 50 Millionen Euro nicht überschreitet [13]. Es werden zehn Jahre Indikationsschutz für das erste als Orphan Drug zugelassene Arzneimittel garantiert. Dies bedeutet, dass ein Produkt eines anderen Herstellers während dieser Zeit nicht zugelassen werden kann.
wirtschaftliche Anreize |
indirekte wirtschaftliche Anreize |
|---|---|
Marktexklusivität |
wissenschaftliche Beratung durch die EMA bzw. das CHMP |
nationale Förderprogramme |
(meist) keine frühe Nutzenbewertung nach AMNOG |
vergünstigte Zulassungsgebühren |
verkürztes Zulassungsverfahren |
Zulassung von Orphan Drugs
Damit nach der Orphan Regulation ein Orphan Drug zugelassen werden kann, muss für den Wirkstoff ein „wesentlicher Beitrag zur Patientenversorgung“ nachgewiesen werden. Im Englischen spricht man von der Major Contribution to Patient Care. Der Nutzen dieses Medikaments muss für den Patienten spürbar sein, die Lebensqualität muss durch dieses Arzneimittel maßgeblich verbessert werden. Dies wäre auch gewährleistet, wenn die Intervention deutlich besser verträglich ist. Da spezifische Behandlungsmethoden bei einer seltenen Erkrankung fehlen, ist in den meisten Fällen dieser wesentliche Beitrag zur Patientenversorgung bereits durch die Voraussetzungen der Seltenheit des Leidens gegeben.
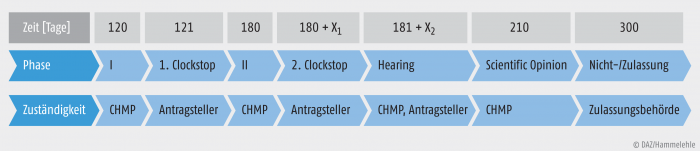
Orphan Drugs müssen innerhalb des EU-Raumes zentral zugelassen werden. Während dieses Zulassungsverfahrens gibt es verschiedene Phasen (Abb. 2). Zu Beginn, während der 1. Bewertungsphase, wird das gesamte Zulassungsdossier dem Committee for Medicinal Products for Human Use (CHMP) der EMA vorgelegt und die Zulassung beantragt. Es werden die regulatorischen Behörden aus zwei Länder als Rapporteur und Co-Rapporteur bestimmt. Diese sind federführend für das Verfahren und erstellen einen ersten Bericht. Rapporteur und Co-Rapporteur haben zunächst 80 Tage Zeit, Beanstandungen an den vorgelegten Unterlagen zu äußern oder fehlende Informationen vom pharmazeutischen Unternehmer einzufordern. Anschließend können alle anderen EU-Mitgliedstaaten 40 Tage lang die Stellungnahme von Rapporteur und Co-Rapporteur kommentieren. Im Zuge des Verfahrens wird eine wissenschaftliche Stellungnahme erstellt, ein Assessment Report. Der Antragsteller erhält danach sechs Monate Zeit, um Informationen nachzureichen oder zu bearbeiten, falls nötig, wird diese Frist verlängert. Während dieser Zeit läuft kein aktiver Bewertungsprozess seitens der Zulassungsbehörden, deshalb wird von einem Clockstop gesprochen. Dies bedeutet, dass die Zeit des Bewertungsverfahrens „angehalten“ wird und die Gesamtzeit des Zulassungsverfahrens von 210 Tagen (inklusive nationaler Phase 300 Tage) dadurch nicht beeinflusst wird.
Abb. 2: Das zentrale Zulassungsverfahren der EU X ist eine undefinierte Zeit. Die Dauer des Verfahrens wird um weitere 90 Tage verlängert, um die informativen Texte in der jeweiligen Landessprache zu prüfen.
In der 2. Phase hat das CHMP weitere 60 Tage, um offene Punkte zu identifizieren. Der Antragsteller kann fehlende Belege innerhalb des 2. Clockstop binnen 30 Tagen nachreichen. Nach dem 181. Tag des Zulassungsverfahrens kommt es zur Anhörung des Antragstellers (Hearing) durch das CHMP. Alle vorliegenden Informationen wie der Zulassungsantrag und die Stellungnahme von Rapporteur und Co-Rapporteur werden zur Entscheidungsfindung mit einbezogen. Eine endgültige Zulassung oder Ablehnung des Antrages wird von der Europäischen Kommission ausgesprochen. Wenn eine zentrale Zulassung erteilt wird, muss ein neuer Bewertungsreport zusammengestellt werden, der European Public Assessment Report (EPAR). Dieser wird der allgemeinen Öffentlichkeit zur Verfügung gestellt. Nach dem 210. Tag der Zulassung schließt sich die Übersetzung der informativen Texte in die jeweilige Amtssprache der EU-Mitgliedsländer an. Für ein zentrales Zulassungsverfahren (Centralized Procedure) werden insgesamt 300 Tage veranschlagt [2]. Grundsätzlich sind für eine zentrale Marktzulassung verlässliche Daten unabdingbar. Diese Unterlagen können nicht immer vollständig oder statistisch ausreichend belastbar sein, da es bei Orphan Drugs oft schwierig ist, eine statistische Aussagekraft in klinischen Studien zu belegen. Daher gibt es zwei besondere regulatorische Optionen im zentralen Zulassungsverfahren:
- die Marktzulassung unter Beauflagung oder
- eine Marktzulassung, die unter bestimmten Voraussetzungen gewährt werden kann. Hierbei müssen nicht wie üblich pivotale klinische Daten vorgelegt werden, diese können nachgereicht werden [2].
Bei der ersten Möglichkeit, der Conditional Marketing Authorisation, wird ein dringend benötigtes Arzneimittel zugelassen, dessen vorläufige Nutzen-Risiko-Abwägung positiv ausfällt. Klinische Daten sollten weiterhin erhoben werden. Wenn es zur Erfüllung der Auflagen kommt (z. B. die klinische Datenlage Signifikanz-Niveau erreicht), wird eine „normale“ zentrale Zulassung erteilt.
Im Gegensatz dazu kommt es bei der älteren Option Marketing Authorisation under Exceptional Circumstances selten zur Vervollständigung des Dossiers (Artikel 14(8) EG/726/2004) [6]. Es handelt sich um Zulassungen in Ausnahmefällen, wenn es keine Möglichkeiten zur pivotalen klinischen Datenerhebung gibt. Das ist z. B. der Fall, wenn auch bei einem langfristigen Follow-up nicht zu erwarten ist, dass in Phase III der klinischen Entwicklung statistische Signifikanz erreicht wird. Beide Möglichkeiten der Zulassung stellen Ausnahmen dar.
Unterschiede von Orphan Designation und Marketing Authorisation
Die Ausweisung des Arzneimittels für eine seltene Erkrankung (Orphan Designation) kann nicht mit einer Marktzulassung gleichgesetzt werden (siehe Tabelle 2). Denn bei einer Orphan Designation wird lediglich während des laufenden Arzneimittel-Entwicklungsprogramms die Anerkennung erteilt, dass es sich um einen Wirkstoff gegen seltene Leiden handelt. Damit sollen die anfangs erwähnten Anreize geschaffen werden, um die Entwicklung von Arzneimitteln gegen seltene Leiden zu fördern. Bei der Marktzulassung hingegen soll sichergestellt werden, dass nur Arzneimittel, die wirksam, sicher und von hoher Qualität sind, vermarktet werden. Generell ist für Orphan Drugs das gleiche Maß an Evidenz im Hinblick auf das Risiko nötig.
Designation |
Marktzulassung |
|
|---|---|---|
wer? |
natürliche oder juristische Person |
nur pharmazeutische Unternehmen |
wann? |
während der Entwicklung vor Marktzulassung |
nach der klinischen Entwicklung |
Grad der Evidenz |
angemessene wissenschaftliche Begründung |
wissenschaftlich bestätigte Fakten |
Rechtlicher Rahmen der Orphan Drugs
Damit die Vorteile eines Arzneimittels gegen seltene Erkrankungen nur denjenigen Unternehmen zugute kommen, welche auf seltene Indikationen hinarbeiten, ist ein geeigneter rechtlicher Rahmen erforderlich. Die Orphan Regulation EG/141/2000 erfüllt den Zweck, ein europäisch zentrales Verfahren für Arzneimittel gegen seltene Leiden zu schaffen und Anreize für die Erforschung, Entwicklung und das Inverkehrbringen von Orphan Drugs zu setzen [1].
Einzelheiten, Definitionen und Fragestellungen rund um die Marktexklusivität klärt ergänzend zur Orphan Regulation die Verordnung EG/847/2000 [5]. In dieser Verordnung wird zunächst der Begriff des Arzneimittels gegen seltene Leiden geklärt. Es werden weitere Begriffe definiert, wie beispielsweise ähnliches Arzneimittel (Similar Medicinal Product) und klinische Überlegenheit (Clinical Superiority). Diese Abgrenzung ist notwendig, wenn ein weiteres Arzneimittel den gleichen Markt beansprucht (Similarity Assessments), was bei Zulassungen von mehreren Orphan Drugs innerhalb einer Indikation der Fall sein kann (siehe hierzu auch den Artikel „Ähnlich, gleich oder klinisch überlegen? Das Similarity Assessment bei Orphan Drugs“, S. 66) [5].
Zusätzliche Vorteile ergeben sich, wenn ein Orphan Drug bei Marktzulassung einen Pedeatric Investigation Plan aufweist, dies wird in der Pediatric Regulation EG/1901/2006 erläutert. Als Beispiel dient das Nephroblastom (Wilms-Tumor), als eine Erkrankung, die selten und (fast) ausschließlich bei Kindern vorkommt. Die allgemeine rechtliche Grundlage zur zentralisierten Marktzulassung eines Orphan Drugs bietet die EG/726/2004, die jeweiligen Zuständigkeiten der Behörden und Gremien werden ebenfalls geregelt [6]. Die Nutzung der Conditional Marketing Authorisation für Orphan Drugs wird in der Verordnung EG/507/2006 ausgeführt.
Ausblick
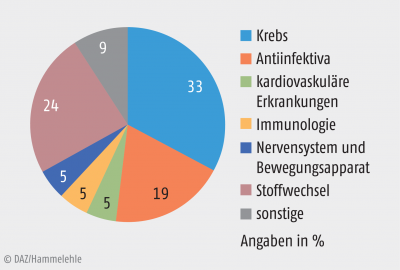
Tabelle 3 zeigt die bisher zugelassenen 21 Orphan Drugs der Jahre 2014 und 2015 [8]. Die Indikationen lassen ein Muster erkennen: Es wurden relativ viele Onkologika zugelassen, sowie Antiinfektiva (Tuberkulosetherapeutika) und Arzneimittel für ungewöhnliche Erkrankungen wie Morbus Cushing (siehe Abb. 3). Derzeit umfassen die zugelassenen Arzneimittel gegen seltene Leiden ungefähr 30 Indikationen [8]. Trotz der Anreize für die pharmazeutische Industrie werden bei Weitem noch nicht alle benötigten Indikationen abgedeckt. Die Europäische Kommission geht davon aus, dass es 5000 bis 8000 seltene Leiden gibt [10].
Arzneimittel |
Wirkstoff |
Zulassungsdatum |
Indikation |
Einteilung |
|---|---|---|---|---|
|
Tasimelteon®
|
Hetlioz |
7. Juli 2015 |
Behandlung von Schlaf-Wach-Störungen mit Abweichungen vom 24-Stunden-Rhythmus bei blinden Personen ohne Lichtwahrnehmung |
sonstige |
|
Lenvatinib®
|
Lenvima |
1. Juni 2015 |
Behandlung von follikulärem und papillärem Schilddrüsenkarzinom |
Krebs |
|
Holoclar®
|
(autologes menschliches Corneaepithel mit Stammzellen) |
19. Februar 2015 |
Behandlung von Hornhauterkrankungen |
sonstige |
|
Cerdelga®
|
Eliglustat |
21. Januar 2015 |
Langzeitbehandlung Morbus Gaucher Typ 1 |
Stoffwechsel |
|
Ofev®
|
Nintedanib |
19. Januar 2015 |
Behandlung der idiopathischen Lungenfibrose (IPF) |
Antiinfektiva |
|
Scenesse®
|
Afamelanotid |
29. Dezember 2014 |
Behandlung von erythropoetischer Protoporphyrie (EPP) |
Stoffwechsel |
|
Cyramza®
|
Ramucirumab |
23. Dezember 2014 |
Behandlung von Magenkarzinom |
Krebs |
|
Lynparza®
|
Olaparib |
18. Dezember 2014 |
Therapie von Ovarialkarzinom |
Krebs |
|
Ketoconazole HRA®
|
Ketoconazol |
21. November 2014 |
Behandlung des Cushing-Syndroms |
Stoffwechsel |
Imbruvica |
Ibrutinib |
23. Oktober 2014 |
Behandlung von chronisch lymphatischer Leukämie (CLL) und Mantelzell-Lymphom (MCL) |
Krebs |
|
Arzerra®
|
Ofatumumab |
21. April 2010 [Indikationserweiterung: 5. August 2014] |
Behandlung von CLL [bei zuvor unbehandelten Patienten] |
Krebs |
|
Translarna®
|
Ataluren |
5. August 2014 |
Behandlung der Duchenne-Muskeldystrophie |
Nervensystem und Bewegungsapparat |
|
Gazyvaro®
|
Obinutuzumab |
24. Juli 2014 |
Behandlung von CLL |
Krebs |
|
Sylvant®
|
Siltuximab |
27. Mai 2014 |
Behandlung der Castleman-Krankheit |
Immunologie |
|
Vimizim®
|
Elosulfase alfa |
30. April 2014 |
Behandlung von Mucopolysaccharidose, Typ IVA |
Stoffwechsel |
|
Deltyba®
|
Delamanid |
30. April 2014 |
Behandlung von Tuberkulose |
Antiinfektiva |
|
Granupas®
|
Paraaminosalicylsäure (PAS) |
09. April 2014 |
Behandlung von Tuberkulose |
Antiinfektiva |
|
Kolbam®
|
Cholsäure |
08. April 2014 |
Behandlung angeborener Störungen der Synthese primärer Gallensäuren |
Stoffwechsel |
|
Adempas®
|
Riociguat |
31. März 2014 |
Behandlung von pulmonaler arterieller Hypertonie (PAH), einschließlich chronischer thromboembolischer pulmonaler Hypertonie |
kardiovaskuläre Erkrankungen |
|
Cometriq®
|
Cabozantinib |
26. März 2014 |
Behandlung von medullärem Schilddrüsenkarzinom |
Krebs |
|
Sirturo®
|
Bedaquilin |
7. März 2014 |
Behandlung von Tuberkulose |
Antiinfektiva |
Da bisher nur die Industrienationen (z. B. USA, Japan, Europa, Singapur, Australien) eine Orphan-Drug-Gesetzgebung implementiert haben und große Teile der Welt von der Versorgung mit wichtigen Orphan Drugs nicht profitieren, wäre eine globale Strategie sicherlich wünschenswert.
Abb. 3: Indikationsgebiete der zugelassenen Orphan Drugs 2014/2015.
Auch würde durch mehr Patienten in einer Indikation (höhere Fallzahlen) die benötigten Arzneimittel mit höherer klinischen Evidenz entwickelt werden. Durch eine veränderte Gesamtpopulation wäre die Prävalenz weltweit betrachtet auch anders verteilt im Vergleich zu lokal eingegrenzten Populationen. In diesem Zusammenhang kann die Tuberkulose als eines von vielen Beispielen dienen (allein 2014 wurden drei Orphan Drugs in dieser Indikation zugelassen). Diese Krankheit ist in der Europäischen Union selten, andernorts jedoch eine erschreckend häufige, sehr reale Bedrohung.
Zudem ist es gerade bei der geringen Population der Orphan Indikationen wichtig, dass Ärzte und medizinisches Fachpersonal auch auf seltene Leiden sensibilisiert werden und somit frühzeitig mit individuellen Therapien helfen können. |
Literatur
[1] Europäisches Parlament (16.12.1999): Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates über Arzneimittel für seltene Leiden
[2] Eckstein N. Arzneimittel - Entwicklung und Zulassung. Für Studium und Praxis. 1. Auflage: Deutscher Apotheker Verlag Stuttgart 2013
[3] Enzmann H, Lütz J. Förderung von Arzneimitteln für seltene Leiden durch die Europäische Gemeinschaft. Springer Medizin Verlag, 2008;51:500–508
[4] eCFR - Orphan Drugs, 21 eCFR 1.316.2013; electronic Code of Federal Regulations (eCFR): Title 21 Food and Drugs, Chapter I Food and Drug Administration, Department of Health and Human Services, Subchapter D Drugs for Human Use, Part 316: Orphan Drugs; current as of 5. August 2015
[5] Verordnung (EG) Nr. 847/2000 zur Festlegung von Bestimmungen für die Anwendung der Kriterien für die Ausweisung eines Arzneimittels als Arzneimittel für seltene Leiden und von Definitionen für die Begriffe „ähnliches Arzneimittel“ und „klinische Überlegenheit“, Europäische Kommission, 27. April 2000
[6] Verordnung (EG) Nr. 726/2004 zur Festlegung von Gemeinschaftsverfahren für die Genehmigung und Überwachung von Human- und Tierarzneimitteln und zur Errichtung einer Europäischen Arzneimittel-Agentur. Europäisches Parlament (30. April 2004): Artikel 14(8) EG/726/2004
[7] The Committee for Orphan Medicinal Products (COMP), The European Medicines Agency; www.ema.europa.eu, Zugriff 12. August 2015
[8] Pharmaceuticals-Community Register. Register of designated Orphan Medicinal Products (by number). Hrsg. European Commission, Zugriff 7. August 2015
[9] Guideline on Procedures for the granting of a marketing authorisation under exceptional circumstances, pursuant to Article 14 (8) of Regulation (EC) NO 726/2004, Committee for Medicinal Products for Human Use (2005), Hrsg. European Medicines Agency
[10] Significant benefit of orphan drugs: concepts and future developments, European Medicines Agency 2012
[11] Latest Orphan Medicinal Products. European Commission, www.ec.europa.eu, Zugriff 4. August 2015
[12] Erstellung und Einreichung eines Dossiers zur Nutzenbewertung gemäß § 35a SGB V. Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards, Gemeinsamer Bundesausschuss (G-BA) 2013, www.g-ba.de
Autoren
Vladlena Pfeifer studiert pharmazeutische Chemie und verbringt die Praxisphase ihres Studiums bei Prof. Dr. Niels Eckstein im Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM).
Anja Metzger hat angewandte Pharmazie an der Hochschule Kaiserslautern studiert und schreibt ihre Abschlussarbeit bei Dr. Paesler und Prof. Eckstein am BfArM.
Martin Norta ist Apotheker, verfügt über viele Jahre Erfahrung im regulatorischen Bereich und leitet im BfArM das Fachgebiet 21 (Verfahrensmanagement/Drug Regulatory Affairs).
Priv.-Doz. Dr. Harald Enzmann ist habilitierter Mediziner und langjährig erfahrener Wissenschaftler im onkologischen Bereich. Er leitet die Abteilung 2 Zulassung im BfArM und ist Mitglied im Ausschuss für Humanarzneimittel der EMA (CHMP).
Prof. Dr. Niels Eckstein ist Wissenschaftler am Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und Professor für Regulatory Affairs und Pharmakologie am Campus Pirmasens der FH Kaiserslautern.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.