
















- DAZ.online
- DAZ / AZ
- DAZ 4/2023
- Cave bei CBD und ...

Foto: chalermchai/AdobeStock
Recht
Cave bei CBD und Vaporisatoren
Welche rechtlichen Fallstricke bei der Abgabe in Apotheken lauern
Außer in Form von Arzneimitteln werden CBD und CBD-haltige Zubereitungen in Nahrungsergänzungsmitteln in den Verkehr gebracht. Nahrungsergänzungsmittel sollen ernährungsbedingte Mängel ausgleichen, eine geeignete Zufuhr bestimmter Nährstoffe aufrechterhalten oder bestimmte physiologische Funktionen unterstützen. Sie sind demnach keine Arzneimittel und erzielen keine pharmakologische, immunologische oder metabolische Wirkung. Dennoch werden CBD-haltige Nahrungsergänzungsmittel in Deutschland vereinzelt von Ärzten mit grünem Rezept „verordnet“ (s. Abbildung 1) und von Apotheken basierend auf einer solchen Empfehlung oder auch eigeninitiativ abgegeben; nicht selten fragen Patienten, ausgehend von Informationen aus dem Internet, in Apotheken auch gezielt nach solchen Produkten als Substitution für (wesentlich teurere) CBD-basierte Arzneimittel. Bei der Inverkehrbringung von CBD-basierten Nahrungsergänzungsmitteln ergeben sich einige Spannungsfelder [1], denen häufig nur unzureichend Rechnung getragen wird. So bewegen sich gesundheitsbezogene Aussagen und Aussagen zur Reduzierung eines Krankheitsrisikos für CBD-haltige Nahrungsergänzungsmittel in Europa zumindest in einer Grauzone, und es ist zweifelhaft, ob diesbezügliche Werbung erlaubt ist.
THC-Grenzwerte bei CBD-Produkten häufig überschritten
Bei der Sicherheitsbeurteilung solcher Produkte hinsichtlich des THC-Gehalts ist keinesfalls der betäubungsmittelrechtliche Gehalt von max. 0,2% THC maßgeblich (s. Abbildung 1), sondern die von der Europäischen Behörde für Lebensmittelsicherheit (EFSA) festgelegte akute Referenzdosis (ARfD) von 1 μg/kg KG/Tag. Nahrungsergänzungsmittel aus Cannabis mit THC-Gehalten, die bereits im Bereich des „Lowest Observed Adverse Effect Level“ (LOAEL) von über 2,5 mg/Tag liegen (unter Berücksichtigung der vorhersehbaren Verzehrmenge), sind als gesundheitsschädlich im Sinne von Art. 14 Abs. 2a i. V. m. Abs. 4 der Verordnung (EG) Nr. 178/2002 zu beurteilen. Wie Untersuchungen der lebensmittelüberwachenden Behörden zeigen, werden diese Grenzwerte bei Cannabis-Produkten, die als Nahrungsergänzungsmittel vermarktet werden, häufig überschritten [2].
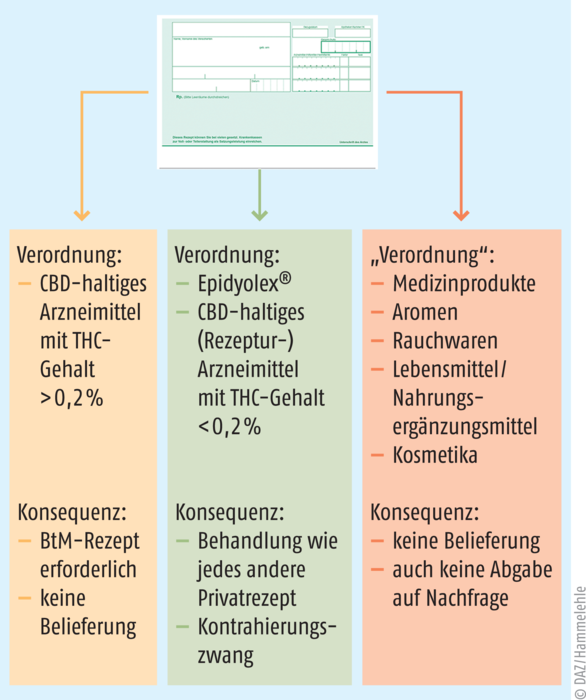
Abb. 1: Immer wieder kommen Patienten mit grünen Rezepten in die Apotheke, auf denen Cannabis(-Zubereitungen) stehen. Das Schema zeigt, unter welchen Bedingungen derlei Verordnungen rechtskonform beliefert werden können.
CBD-Produkte als Novel Food zulassungspflichtig
Sogenannte „Neuartige Lebensmittel“ müssen in der EU zugelassen werden, um verkehrsfähig zu sein. Zwischen den EU-Mitgliedstaaten abgestimmte Informationen über den Novel-Food-Status vieler Lebensmittel stehen im sog. „Novel Food Katalog“ der EU. Extrakte aus Cannabis sativa L. und daraus hergestellte Produkte, die Cannabinoide enthalten, werden in der EU als neuartige Lebensmittel betrachtet, da sie vor dem Stichtag am 15. Mai 1997 nicht in nennenswertem Umfang in der Europäischen Union für den menschlichen Verzehr verwendet wurden. Sie wurden in den „Novel Food Katalog“ aufgenommen. Dies gilt sowohl für die Extrakte selbst, als auch für alle Produkte, denen sie als Zutat zugesetzt werden (z. B. Hanfsamenöl), sowie für Extrakte aus anderen Pflanzen, die Cannabinoide enthalten. Synthetisch gewonnene Cannabinoide werden ebenfalls als neuartig betrachtet. Auch wenn der „Novel Food Katalog“ nicht rechtsverbindlich ist, stellt er dennoch ein zwischen den Mitgliedstaaten abgestimmtes, quasi bindendes Dokument dar. Im Rahmen gerichtlicher Auseinandersetzungen hat er eine Indizwirkung, was bedeutet, dass die betroffenen Unternehmen belegbare Fakten über den Verzehr ihres Produkts vor dem Stichtag vorlegen müssten, um nachzuweisen, dass es nicht unter die Novel-Food-Verordnung fällt. Es ist deshalb davon auszugehen, dass innerhalb der EU alle Extrakte und Zubereitungen aus Cannabis (außer aus Samen) und isolierte Cannabinoide sowie alle Produkte, denen Cannabinoide zugesetzt wurden, einen Novel-Food-Status haben und deshalb ohne Zulassung nicht verkehrsfähig sind. Bisher liegen diesbezügliche Zulassungen nicht vor, wenngleich eine Reihe von Anträgen bei der EFSA eingereicht wurden.
Laut EFSA viele Fragen offen
Mitte des Jahres 2022 publizierte die EFSA eine umfangreiche Stellungnahme zur Sicherheitsbewertung von Cannabidiol (CBD) in (neuartigen) Lebensmitteln und die bei der Bewertung identifizierten Datenlücken [3]. Diese Lücken betreffen die ADME- (Absorption, Distribution, Metabolismus und Elimination) Charakteristika von CBD, das Interaktionspotenzial mit Arzneimitteln sowie die Sicherheit der Einnahme von CBD als Lebens- oder Nahrungsergänzungsmittel. Auch eine mögliche toxikologische Wirkung von CBD auf die Leber, den Magen-Darm-Trakt, das endokrine System, das Nervensystem und die Psyche muss aus Sicht der EFSA noch geklärt werden. Studien an Tieren zeigen eine signifikante Reproduktionstoxizität; inwieweit diese beim Menschen im Allgemeinen und bei Frauen im gebärfähigen Alter im Besonderen auftritt, muss ebenfalls noch untersucht werden. Solange die Antragsteller keine Daten vorlegen, die geeignet sind, diese Lücken zu schließen, wird die EFSA die vorliegenden Anträge für CBD-haltige neuartige Lebensmittel nicht weiterbearbeiten, so bleiben CBD-haltige Nahrungsergänzungsmittel bis auf Weiteres nicht verkehrsfähig.
Apotheken haften für unrechtmäßig in Verkehr gebrachte CBD-Produkte
Unternehmen, die CBD-Produkte als Nahrungsergänzungsmittel in Verkehr bringen, werben dennoch häufig mit abenteuerlichen Begründungen, warum ihre Produkte nicht als Novel Food anzusehen und deshalb verkehrsfähig sein sollen. Derartigen Ausarbeitungen sollte man mit großer Skepsis begegnen. Ein schönes Beispiel dafür ist die Bezugnahme auf „natürliche“ CBD-Gehalte, wie sie in Pflanzen vorkommen, ohne dabei zu berücksichtigen, dass es keine Datenbasis für solche „natürlichen“ CBD-Gehalte in der historischen Verwendung von Hanfprodukten gibt und dass Extrakte eine ganz andere Matrix im Vergleich zu historisch verwendeten Hanfprodukten abbilden. Außerdem ist eine solche Ausnahme im „Novel-Food-Katalog“ nicht vorgesehen. Dort werden Extrakte unisono als neu kategorisiert. Das wurde auch seitens des Bundesamts für Verbraucherschutz und Lebensmittelsicherheit klargestellt [4]: „Dem Bundesamt für Verbraucherschutz und Lebensmittelsicherheit ist derzeit keine Fallgestaltung bekannt, wonach der Verkauf von CBD-Öl erlaubt wäre.“ Zum gleichen Schluss kommen auch die Autoren einer wissenschaftlichen Ausarbeitung für den Deutschen Bundestag [5]. Auch die rezente Rechtsprechung folgt unisono dieser Auslegung, inzwischen auch in zweiter Instanz. Dennoch ergibt sich die paradoxe Situation, dass diese Auffassung nicht stringent von den zuständigen Überwachungsbehörden umgesetzt wird. Das BVL weist darauf hin, dass die Einstufung von Erzeugnissen und die Bewertung der Verkehrsfähigkeit Aufgabe der für die Lebensmittelüberwachung zuständigen Landesbehörden sei. Die Auffassung des BVL zur Verkehrsfähigkeit könne daher nur vorbehaltlich einer abweichenden Ansicht der jeweils zuständigen Überwachungsbehörden in den Bundesländern gelten. Dabei erscheint es wichtig festzuhalten, dass lebensmittelrechtlich Inverkehrbringer nicht nur die Hersteller sind, sondern auch Großhändler und Apotheken, die Produkte an Endverbraucher abgeben. In diesem Zusammenhang sei erwähnt, dass das Inverkehrbringen derartiger Produkte ohne Novel-Food-Zulassung strafbar ist (§ 1a NLV iVm § 59 Abs. 3 Nr. 2a LFGB).
Klassifizierung als Präsentations- oder Funktionsarzneimittel
Ein weiteres Problem ergibt sich aus dem Umstand, dass einschlägige CBD-Produkte auch als Präsentations- oder Funktionsarzneimittel aufgefasst werden können. In jüngster Zeit gab es bereits Fälle, in denen das BfArM basierend auf § 21 Abs. 4 AMG nach Anfragen der zuständigen Landesbehörden CBD-Produkte als Funktionsarzneimittel klassifiziert hat. Solche Entscheidungen binden nicht nur die anfragenden Landesbehörden, sondern auch andere Behörden und Zivilgerichte. Im vorliegenden Fall wurde die Entscheidung des BfArM angefochten. Das Verwaltungsgericht Köln (Urteil vom 22. März 2022 – 7 K 954/20) hat die Einstufung des BfArM bestätigt – im Kern mit der Begründung, dass es sich bei CBD um einen pharmakologisch aktiven Wirkstoff handle, es dazu mit Epidyolex ein zugelassenes Arzneimittel gibt und dass trotz der gegenüber Epidyolex geringeren Dosis von einer pharmakologischen Wirkung auszugehen sei. Dem Produkt fehle zudem „jeder Ernährungszweck“; es handle sich bei CBD also um einen Arzneistoff ohne lebensmittelbezogenen Zweck. Jenseits der Novel-Food-Problematik wird die Verkehrsfähigkeit solcher Produkte damit ganz grundsätzlich infrage gestellt. Gleichzeitig ist mit der Abgabe solcher Produkte wiederum eine Strafbarkeit verbunden (§ 96 Nr. 5 AMG). Insofern ist festzuhalten, dass die Abgabe von CBD-Nahrungsergänzungsmitteln (in der Apotheke) insgesamt mit erheblichen strafrechtlichen Risiken verbunden ist, da auch die Abgabe in der Apotheke als ein Inverkehrbringen entsprechend § 4 Abs. 17 AMG zu betrachten ist.
Vaporisatoren sind zertifizierungspflichtige Medizinprodukte
Medizinisches Cannabis (Blüten oder Extraktzubereitungen) wird häufig durch Vaporisation appliziert. Nach Artikel 1 (9) der Europäischen Verordnung zu Medizinprodukten („Medical Device Regulation“, MDR) sind die dabei verwendeten Verdampfer als Medizinprodukte zu kategorisieren. Die Klassifizierung basiert auf den in Anhang VIII der MDR festgelegten Kriterien. Verdampfer von Arzneimitteln werden in die Klasse IIb eingestuft [7]. Für solche Produkte muss die Konformitätsbewertung grundsätzlich unter Einbeziehung einer EU-Benannten Stelle durchgeführt werden. Darüber hinaus erfordert die Risikoklasse IIb klinische Studien mit diesen Medizinprodukten (und spezifischen Zubereitungen). Verdampfer, die mit der Zweckbestimmung abgegeben (oder in Verkehr gebracht) werden, medizinisches Cannabis zu verabreichen und nicht zertifiziert sind, dürfen nach § 93 Abs. 3 MPDG nicht in Betrieb genommen werden und deren Inverkehrbringung kann behördlicherseits untersagt werden. Auch in diesem Fall kann sich ein Strafbarkeitsrisiko für die abgebende Apotheke ergeben.
2024: Alle Vaporisatoren müssen Konformitätsbewertungsverfahren durchlaufen haben
Der Nachweis der Konformität liegt in der Verantwortung des Herstellers des Medizinprodukts. In jedem Fall werden die allgemeinen Sicherheits- und Leistungsanforderungen, die klinische Leistung und die technische Dokumentation von der Benannten Stelle bewertet. In Falle von Verdampfern, die generisch – also zur Applikation unterschiedlicher Cannabis-Arzneimittel – in den Verkehr gebracht werden, ist noch ungeklärt, wie die Konformitätsbewertung erfolgen kann, da dazu zubereitungsspezifische (klinische) Daten nötig sind. Die Produkte müssen zukünftig außerdem mit einer UDI-Nummer (Unique Device Identifier) versehen und im elektronischen System EUDAMED gemäß Artikel 29 der MDR registriert werden. Bisher sind in Europa keine Verdampfer verfügbar, deren Konformität entsprechend der MDR belegt wurden und die ein entsprechendes CE-Signum tragen. Die als Medizinprodukte nach den bisherigen Regelungen ohne Konformitätsbewertung durch eine Benannte Stelle zertifizierten Verdampfer der Firma Storz & Bickel können im Rahmen einer Übergangsregelung noch bis Mai 2024 in den Verkehr gebracht werden und in Apotheken mit medizinischem Cannabis abgegeben werden. Die Kosten werden auf Antrag auch von den Kassen erstattet. Darüber hinaus existieren zurzeit noch keine anderen zertifizierten Verdampfer. Häufig versuchen Hersteller solcher nicht zertifizierter Geräte mit CE-Kennzeichnungen im Kontext anderer Vorgaben eine EU-Zertifizierung als Medizinprodukt vorzutäuschen.
Prüfpflichten der Apotheke als Medizinproduktehändler bzw. -betreiber
Bevor Apotheken ein bestimmtes Medizinprodukt auf dem Markt bereitstellen, müssen sie nach Artikel 14 der MDR überprüfen, ob alle folgenden Anforderungen erfüllt sind:
- Das Produkt trägt die CE-Kennzeichnung, und es wurde eine EU-Konformitätserklärung für das Produkt ausgestellt (das erfordert eine formale Prüfung, ob das Medizinprodukt ein CE-Signum trägt das auf einer Konformitätsbewertung nach der MDR beruht; zukünftig findet man diese Informationen in der EUDAMED-Datenbank, sie sollten auf jeden Fall vom Hersteller bereitgestellt werden);
- dem Produkt liegen die vom Hersteller gemäß Art. 10 Abs. 11 bereitgestellten Informationen bei (dabei handelt es sich um Informationen in deutscher Sprache; sachliche, nicht jedoch fachliche Prüfung, ob durch den Hersteller nach Art. 10 Abs. 11 MDR bereitzustellende Informationen in deutscher Sprache, u. a. Gebrauchsanweisung, Zweckbestimmung, Seriennummer, Restrisiken, Kontraindikationen und alle unerwünschten Nebenwirkungen etc.);
- bei importierten Produkten hat der Importeur die in Art. 13 Abs. 3 genannten Anforderungen erfüllt (Importeure geben auf dem Produkt oder auf seiner Verpackung oder auf einem dem Produkt beiliegenden Dokument ihren Namen, ihren eingetragenen Handelsnamen oder ihre eingetragene Handelsmarke, ihre eingetragene Niederlassung und die Anschrift an, unter der sie zu erreichen sind, sodass ihr tatsächlicher Standort ermittelt werden kann. Sie sorgen dafür, dass eine zusätzliche Kennzeichnung die Informationen auf der vom Hersteller angebrachten Kennzeichnung nicht verdeckt.);
- gegebenenfalls wurde vom Hersteller eine UDI vergeben (der Unique Device Identifier (UDI) ist ein eindeutiger (alpha)numerischer Code für ein Medizinprodukt. Er ermöglicht eine eindeutige Identifizierung, erleichtert Rückverfolgbarkeit, Rückruf sowie die Bekämpfung von Fälschungen und verbessert die Patientensicherheit).
Meldepflichten der Apotheke als Medizinproduktehändler bzw. -betreiber
Die Medizinprodukte-Anwendermelde- und Informationsverordnung (MPSV) ermöglicht es den zuständigen Bundesoberbehörden auftretende Vorkommnisse zu erfassen, zu bewerten und je nach Risiko Maßnahmen einzuleiten. Meldepflichtig sind alle, die Medizinprodukte beruflich/gewerblich an Patienten/Laien, die diese Medizinprodukte in Eigenanwendung nutzen, abgeben und im Rahmen dessen von einem Vorkommnis erfahren sowie alle, die Medizinprodukte beruflich oder gewerblich betreiben oder anwenden. Vorkommnisse sind:
- Funktionsstörungen,
- Ausfall des Medizinprodukts,
- Änderung der Merkmale,
- Änderung der Leistung,
- unsachgemäße Kennzeichnung,
- unsachgemäße Gebrauchsanweisung,
- Mangel an Gebrauchstauglichkeit.
Die Meldung erfolgt auf elektronischem Weg über die Homepage des BfArM. Möglicherweise ergeben sich aus der Berufsordnung der jeweiligen Landesapothekerkammer parallele Meldepflichten an die AMK.
Nicht-zertifizierte Vaporisatoren sind keine apothekenüblichen Waren
Verschiedentlich stellen sich Apotheken die Frage, ob es nicht möglich sei, nicht-zertifizierte Vaporisatoren in der Apotheke zu verkaufen, solange diese nicht für die Verabreichung von Arzneimitteln beworben oder gar empfohlen werden, schließlich obläge es in diesem Fall den Kunden, die ein solches Produkt erwerben, wofür diese den Vaporisator nutzen. Eine solche Argumentation scheitert jedoch an § 2 Abs. 4 der Apothekenbetriebsordnung (ApBetrO), derzufolge der Apothekenleiter neben Arzneimitteln und apothekenpflichtigen Medizinprodukten nur die in § 1a Abs. 10 ApBetrO genannten apothekenüblichen Waren anbieten oder feilhalten darf. Dazu zählen zwar auch nicht-apothekenpflichtige Medizinprodukte, wozu auch die o. g. zertifizierten Vaporisatoren gehören; Vaporisatoren, die nicht als Medizinprodukt zertifiziert sind, unterfallen jedoch keiner der übrigen in § 1a Abs. 10 ApBetrO genannten Produktkategorien. Sie dürfen damit nicht in Apotheken abgegeben werden. Andernfalls könnte man auch auf die Idee kommen Tabakpfeifen oder E-Zigaretten in der Apotheke anzubieten, was die Absurdität eines solches Ansinnens plastisch vor Augen führt. Sollen in Apotheken auch nach 2024 (s. o.) weiterhin Vaporisatoren zum Verdampfen von Cannabis angeboten werden können, führt der Weg demnach nur über zertifizierte Medizinprodukte, die ein Konformitätsbewertungsverfahren nach MDR durchlaufen haben. |
Literatur
[1] Veit, M.; Ziegler, A. CBD-Produkte: Verkehrsfähigkeit und Abgrenzung zu Medizinal-Cannabis. Pharm. Ind. 2022; 84(1):84-97.
[2] Lachenmeier, DW, Walch, SG. Evidence for adverse effects of cannabidiol (CBD) products and their non-conformity on the European food market – response to the European Industrial Hemp Association. F1000Res. 2020;9:1051. doi:10.12688/f1000research.26045.2
[3] EFSA Panel on Nutrition, Novel Foods and Food Allergens (NDA). Statement on safety of cannabidiol as a novel food: datagaps and uncertainties. EFSA Journal 2022;20(6):7322. DOI: https://doi.org/10.2903/j.efsa.2022.7322
[4] BVL. Hanf, THC, Cannabidiol (CBD) & Co. 2021. https://www.bvl.bund.de/DE/Arbeitsbereiche/01_Lebensmittel/04_AntragstellerUnternehmen/13_FAQ/FAQ_Hanf_THC_CBD/FAQ_Cannabidiol_node.html (Zugriff: 02.10.2022).
[5] Deutscher Bundestag – Wissenschaftliche Dienste. Verkehrsfähigkeit von Cannabidiol(CBD)-haltigen nicht medizinischen Produkten in Deutschland: WD 5 – 3000 – 088/20. 2020.
[6] Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies with respect to the implementation of the Medical Devices and In Vitro Diagnostic Medical Devices Regulations ((EU) 2017/745 and (EU) 2017/746) (EMA/37991/2019).
[7] Regel 20 des Anhangs VIII der MDR zzgl. der Erläuterungen in der MDCG 2021-24 „Guidance on classification of medical devices“).
Teile des Beitrags wurden vorab in der Pharmazeutischen Industrie veröffentlicht (Pharm. Ind. 2022;84(12):1368-1377).
Autoren
Prof. Dr. Markus Veit
Pharmaziestudium in Frankfurt, Promotion und Habilitation im Fach Pharmazeutische Biologie an der Julius-Maximilians-Universität in Würzburg, Fachapotheker für Pharmazeutische Analytik und Mitglied im Ausschuss Pharmazeutische Chemie des Arzneibuchs beim BfArM, Geschäftsführer in Dienstleistungsunternehmen für die Pharmazeutische Industrie mit den Schwerpunkten Arzneimittelentwicklung, -prüfung und -zulassung, zur Zeit Geschäftsführer der Alphatopics GmbH
Apotheker Dr. Andreas S. Ziegler, Pharmaziestudium an der Universität Erlangen-Nürnberg; Referent und Wissenschaftsjournalist; Fachapotheker für Pharmazeutische Technologie und Lehrauftrag für Pharmazeutische Technologie an der Uni Erlangen-Nürnberg



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.