














- DAZ.online
- DAZ / AZ
- DAZ 17/2009
- Sapropterin zur ...
Arzneimittel und Therapie
Sapropterin zur Behandlung der Phenylkentonurie
Sapropterin
ist eine synthetische Form des Kofaktors Tetrahydrobiopterin (BH4) der Phenylalaninhydroxylase (PAH) und kann bei Patienten mit PAH-Defekt oder PAH-Mangel die Restaktivität des Enzyms stimulieren, so dass der Phenylalanin-blutspiegel sinkt.
Die Phenylketonurie ist eine angeborene Störung des Aminosäurestoffwechsels, die autosomal-rezessiv vererbt wird und zu abnorm hohen Konzentrationen von Phenylalanin im Blut (Hyperphenylalaninämie) führt.
Die Erkrankung tritt bei einem von 8000 Neugeborenen auf und kann bei Säuglingen und Kleinkindern zu schweren Schädigungen des Gehirns führen, bei älteren Patienten zu neurologischen Schäden. In den Industrienationen gibt es etwa 50.000 Patienten mit einer derartigen Erkrankung.
Das Enzym Phenylalaninhydroxylase fehlt
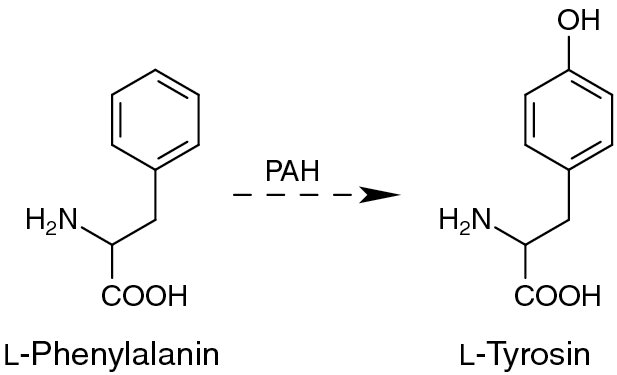
Die Betroffenen können die essenzielle Aminosäure Phenylalanin nicht abbauen, weil ihnen das Enzym Phenylalaninhydroxylase fehlt oder nicht ausreichend aktiv ist. Dieses Enzym überführt Phenylalanin in die Aminosäure Tyrosin.
Tyrosin wird für die Biosynthese des Neurotransmitters Dopamin, der Schilddrüsenhormone, aber auch für die Bildung des Pigmentfarbstoffs Melanin benötigt.
Die Folge des Enzymdefekts sind erhöhte Phenylalaninblutspiegel, und als alternative Abbauprodukte werden die Ketone Phenylpyruvat, Phenylacetat und Phenyllactat vermehrt mit dem Urin ausgeschieden, was der Erkrankung ihren Namen gab.
Ein wichtiges Koenzym der Phenylhydroxylase ist Tetrahydrobiopterin (BH4). Rund 2% der Patienten haben eine atypische Phenylketonurie, bei der der Stoffwechsel dieses Koenzyms gestört ist.
Beeinträchtigung der Hirnentwicklung
Unbehandelt führt der Überschuss an Phenylalanin zu einer Beeinträchtigung der Hirnentwicklung mit meist schweren Störungen der geistigen Entwicklung. Warum die Phenylketonurie die Hirnentwicklung behindert, ist nicht bekannt. Diskutiert wird, ob die erhöhte Menge Phenylalanin andere Aminosäuren im Wettbewerb um die Transportkapazitäten zur Überwindung der Blut-Hirn-Schranke zurückdrängt. Dies könnte einerseits zur Folge haben, dass die körpereigene Proteinbiosynthese im Gehirn gestört wird, und andererseits könnte dadurch die Synthese verschiedener Neurotransmitter beeinträchtigt werden.
Der Mangel an Melanin führt außerdem zu einer auffallend hellen Hautfarbe. Die Kinder sind deshalb häufig hellblond und haben blaue oder rote Augen.
Geschichte der PhenylketonurieIm Jahr 1934 wies der norwegische Arzt Ivar Asbjørn Følling erstmals bei geistig behinderten Patienten eine vermehrte Ausscheidung der Phenylbrenztraubensäure mit dem Urin mit Eisen(III)-chlorid nach ("Fölling-Probe"). 1947 entdeckte dann G. A. Jervis den eigentlichen Defekt in der Umwandlung von Phenylalanin zu Tyrosin. Der nächste Meilenstein war die Einführung einer phenylalaninarmen Diät zur Behandlung der Erkrankung durch den deutschen Kinderarzt Horst Bickel 1953. Zehn Jahre später ermöglichte der amerikanische Mikrobiologe Robert Guthrie mit dem von ihm entwickelten bakteriellen Hemmtest den einfachen Nachweis einer erhöhten Phenylalaninkonzentration im Blut. Die Bestimmung konnte aus Bluttropfen erfolgen, die auf Filterpapier getrocknet wurden, wodurch sich die Methode auch für ein Massenscreening eignete. |
Rechtzeitige Diät verhindert Schäden
Die Erkrankung kann durch eine einfache Reihenuntersuchung schon bei Neugeborenen erkannt werden. Die Symptome können dann durch eine rechtzeitig begonnene eiweißarme Diät mit sehr stark eingeschränkter Aufnahme von Phenylalanin verhindert werden, die lebenslang eingehalten werden sollte.
Normale Phenylalanin-Spiegel im Plasma liegen zwischen 60 und 120 µmol/l (1 bis 2 mg/dl), von einer Hyperphenylalanin-ämie spricht man bei Werten zwischen 120 und 600 µmol/l (2 bis 10 mg/dl). Nach den Empfehlungen der Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (APS) sollten die Plasmaspiegel bis zum 10. Lebensjahr bei 42 bis 240 µmol/l (0,7 bis 4 mg/dl), bis zum 16. Lebensjahr bei 42 bis 900 µmol/l (0,7 bis 15 mg/dl) und danach unter 1200 µmol/l (unter 20 mg/dl) liegen.
Eiweißhaltige Lebensmittel meiden
Da Phenylalanin Bestandteil aller Nahrungseiweiße ist, müssen die Patienten eiweißhaltige Lebensmittel meiden und vor allem auf tierische Lebensmittel wie Fleisch, Fisch, Milch- und Eiprodukte verzichten. Weil auch Aspartam Phenylalanin enthält, müssen kalorienreduzierte Produkte, die mit Aspartam gesüßt wurden, auch immer mit dem Hinweis "enthält eine Phenylalaninquelle" gekennzeichnet werden. Da selbst Weizen und andere Getreideerzeugnisse viel pflanzliches Eiweiß enthalten, gibt es für die Patienten Back- und Teigwaren aus speziellem eiweißarmen Mehl. Um den Mangel an essenziellen Aminosäuren auszugleichen, müssen sie zusätzlich eine spezielle Aminosäuremischung zu sich nehmen.
Nicht alle Patienten sind gleich stark betroffen. Die Aktivität der Phenylalaninhydroxylase ist je nach Art der Mutation unterschiedlich stark eingeschränkt. Deshalb ist die Menge Eiweiß, welche aufgenommen werden kann, ohne dass der Phenylalaninspiegel über den angestrebten Bereich ansteigt, von Patient zu Patient unterschiedlich.
Kofaktor der Phenylalaninhydroxylase
Bisher war in Europa kein Arzneimittel zur Therapie der Phenylketonurie zugelassen.
Sapropterin ist eine synthetische Form des Koenzyms Tetrahydro-
biopterin (6R-BH4).
Der neue Wirkstoff soll bei Patienten mit Phenylketonurie, die sich durch BH4 beeinflussen lässt, die Aktivität der fehlerhaften Phenylalaninhydroxylase erhöhen und dadurch den oxidativen Phenylalaninmetabolismus in ausreichendem Maße steigern oder wiederherstellen.
Sapropterin soll es Betroffenen ermöglichen, eine weitgehend normale Nahrung zu sich zu nehmen. Vor allem Kindern und Jugendlichen, die eine striktere Diät als Erwachsene einhalten müssen, könnte Sapropterin das Leben erleichtern.
Steckbrief: SapropterinHandelsname: Kuvan Hersteller: Merck Pharma GmbH, Darmstadt Einführungsdatum: 1. April 2009 Zusammensetzung: Jede Tablette zur Herstellung einer Lösung zum Einnehmen enthält 100 mg Sapro-pterindihydrochlorid (entsprechend 77 mg Sapropterin). Sonstige Bestandteile: Mannitol (E 421), Calciumhydrogenphosphat, Crospovidon Typ A, Ascorbinsäure (E 300), Natriumstearylfumarat, Riboflavin (E 101). Packungsgrößen, Preise und PZN: 30 Tabletten, 800,22 Euro, PZN 9012789; 120 Tabletten, 3171,95 Euro, PZN 9012795. Stoffklasse: Sonstige Mittel für das alimentäre System und den Stoffwechsel. ATC-Code: A16AX07. Indikation: Sapropterin ist bei Erwachsenen und Kindern ab vier Jahren mit Phenylketonurie zur Behandlung der Hyperphenylalanin-ämie angezeigt, wenn diese nachweislich auf eine solche Therapie ansprechen. Sapropterin ist auch angezeigt zur Behandlung einer Hyperphenylalaninämie bei Patienten mit Tetrahydrobiopterin (BH4)-Mangel, die nachweislich auf eine solche Therapie ansprechen. Dosierung: Sapropterin soll zum Essen als Einzeldosis immer zur gleichen Tageszeit, vorzugsweise morgens in Wasser gelöst eingenommen werden. Die Anfangsdosis von Sapropterin beträgt Patienten mit Phenylketonurie einmal täglich 10 mg/kg Körpergewicht und wird üblicherweise im Bereich von 5 bis 20 mg/kg und Tag eingestellt. Die Anfangsdosis von Sapropterin mit BH4 -Mangel ist einmal täglich 2 bis 5 mg/kg Körpergewicht und kann auf bis zu 20 mg/kg und Tag erhöht werden. Gegenanzeigen: Überempfindlichkeit gegenüber dem Wirkstoff oder einem der sonstigen Bestandteile. Nebenwirkungen: Sehr häufig: Kopfschmerzen, Rhinorrhö. Häufig: laryngopharyngeale Schmerzen, verstopfte Nase, Husten; Durchfall, Erbrechen, Bauchschmerzen; Hypophenylalaninämie. Wechselwirkungen: Auch wenn bisher die gleichzeitige Anwendung von Dihydrofolat-Reduktase-Hemmern (z. B. Methotrexat, Trimethoprim) nicht untersucht worden ist, können diese Arzneimittel möglicherweise den BH4 -Metabolismus beeinflussen. Vorsicht wird empfohlen bei gleichzeitiger Anwendung von Sapropterin mit allen Mitteln, einschließlich solcher, die topisch angewendet werden, die eine Vasodilatation durch Beeinflussung des Stickstoffmonoxid (NO)-Abbaus oder seiner Wirkung hervorrufen, einschließlich klassischer NO-Donatoren, Phosphodiesteraseinhibitoren vom Typ 5 und Minoxidil. Vorsicht ist bei Patienten angezeigt, die gleichzeitig mit Levodopa behandelt werden, da dies zu erhöhter Erregbarkeit und Reizbarkeit führen kann. Warnhinweise und Vorsichtsmaßnahmen: Patienten, die mit Sapro-pterin behandelt werden, müssen weiter eine phenylalaninarme Diät einhalten und sich regelmäßig klinischen Untersuchungen unterziehen. Eine anhaltende oder periodisch auftretende Fehlsteuerung des Phenylalanin-Tyrosin-L-Dihydroxy-phenylalanin (DOPA)-Abbauweges kann zu einer ungenügenden körpereigenen Protein- und Neurotransmittersynthese führen. Im Krankheitsfall wird ein Arztbesuch empfohlen, da die Phenylalaninblutspiegel dann ansteigen können. Bei Patienten mit Neigung zu Krampfanfällen sollte Sapropterin mit Vorsicht angewendet werden. Sapropterin sollte bei Patienten, die gleichzeitig mit Levodopa behandelt werden, mit Vorsicht angewandt werden, da die kombinierte Therapie mit Sapropterin eine erhöhte Erregbarkeit und Reizbarkeit verursachen kann. |
Oral gut wirksam
Sapropterin wird zum Essen als Einzeldosis immer zur gleichen Tageszeit, vorzugsweise morgens, in Wasser gelöst eingenommen. Um den therapeutischen Effekt zu optimieren, kann es erforderlich sein, die Gesamttagesdosis in zwei bis drei Teilmengen über den Tag verteilt einzunehmen.
Nach oraler Einnahme der aufgelösten Tablette wird Sapropterin resorbiert, die maximale Plasmakonzentration (Cmax) wird drei bis vier Stunden nach Nüchterneinnahme erreicht. Sapropterindihydrochlorid wird primär in der Leber zu Dihydrobiopterin und Biopterin verstoffwechselt. Nach oraler Applikation wird es hauptsächlich über die Fäzes und nur eine kleine Menge mit dem Urin ausgeschieden.
Eine Tablette enthält 100 mg Sapropterin. Die Anfangsdosis von Sapropterin beträgt bei Patienten mit Phenylketonurie einmal täglich 10 mg/kg Körpergewicht und wird üblicherweise im Bereich von 5 bis 20 mg/kg und Tag eingestellt. Die Anfangsdosis von Sapropterin mit BH4 -Mangel ist einmal täglich 2 bis 5 mg/kg Körpergewicht und kann auf bis zu 20 mg/kg und Tag erhöht werden.
Als zufriedenstellendes Ansprechen gilt ein Abfall der Phenylalaninblutspiegel um mehr als 30% oder ein vom behandelnden Arzt für einen Patienten individuell definierter therapeutischer Zielwert. Patienten, die dieses Ziel während der beschriebenen einmonatigen Testphase nicht erreichen, müssen als Non-Responder eingestuft werden und sollten keine weitere Therapie mit Sapropterin erhalten.
Nur zwischen 30 und 70% aller Patienten mit Phenylketonurie sprechen auf die Behandlung mit Sapropterin an. Wahrscheinlich ist Sapropterin vor allem dann wirksam, wenn Mutationen in den Kofaktor-Bindungsstellen des Phenylhydroxylase-Gens vorliegen. Ein BH4 -Belastungstest kann Auskunft geben, ob ein Patient auf eine Sapropterinbehandlung anspricht.
In mehreren randomisierten, placebokontrollierte Studien der Phase III bei Patienten mit Phenylketonurie reduzierte Sapropterin die Phenylalaninblutspiegel und erhöhte die Phenylalanintoleranz bei diätetischer Zufuhr.
Nebenwirkungen: Kopfschmerzen und Rhinorrhö
Bei etwa 35% der 579 Patienten, die in klinischen Studien mit Sapropterindihydrochlorid (5 bis 20 mg/kg täglich) behandelt worden sind, traten Nebenwirkungen auf. Am häufigsten waren Kopfschmerzen und Rhinorrhö. Häufig kam es zu laryngopharyngealen Schmerzen, verstopfter Nase und Husten sowie Durchfall, Erbrechen und Bauchschmerzen.
BH4 ist ein Kofaktor der NO-Synthetase. Vorsicht wird empfohlen bei gleichzeitiger Anwendung von Sapropterin mit allen Mitteln, einschließlich solcher, die topisch angewendet werden, die eine Vasodilatation durch Beeinflussung des Stickstoffmon-oxid (NO)-Abbaus oder seiner Wirkung hervorrufen, einschließlich klassischer NO-Donatoren (z. B. Glyceroltrihydrat, Isosorbiddinitrat, Nitroprussidnatrium, Molsidomin), Phosphodiestera-seinhibitoren vom Typ 5 (PDE-5-Inhibitoren) und Minoxidil.
Quelle
Fachinformation von Kuvan® , Stand Dezember 2008.
Presseinformation der Firma Merck, Darmstadt, 26. September 2008.
hel
Phenylalanin
ist eine essenzielle Aminosäure, die im Rahmen der normalen Ernährung aufgenommen wird. Im gesunden Stoffwechsel wird eine geringe Menge des Phenylalanins für die Proteinbiosynthese verwendet, während der große Rest zu Tyrosin hydroxyliert wird. Für die Hydroxylierung ist das Enzym Phenylalaninhydroxylase (PAH) mit dem Kofaktor Tetrahydrobiopterin (BH4) notwendig.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.