













- DAZ.online
- DAZ / AZ
- DAZ 36/2010
- Midostaurin bei Leukämie
Arzneimittel und Therapie
Midostaurin bei Leukämie
Grafik: Uniklinik Magdeburg
Bei der AML vermehren sich unreife weiße Blutkörperchen, die Blasten, unkontrolliert. Über zwei Drittel aller Erwachsenen mit bösartigen Erkrankungen des blutbildenden Systems sind von einer AML betroffen, jährlich erkranken sechs von 100.000 Menschen in Deutschland an dieser Leukämieform. Während bei jüngeren Erwachsenen die Erkrankung in etwa 40% der Fälle durch eine Chemotherapie geheilt werden kann, sprechen ältere Patienten darauf häufig nicht oder nur unzureichend an. Eine Hoffnung der modernen Medizin ruht daher auf zielgerichteten Blutkrebstherapien.
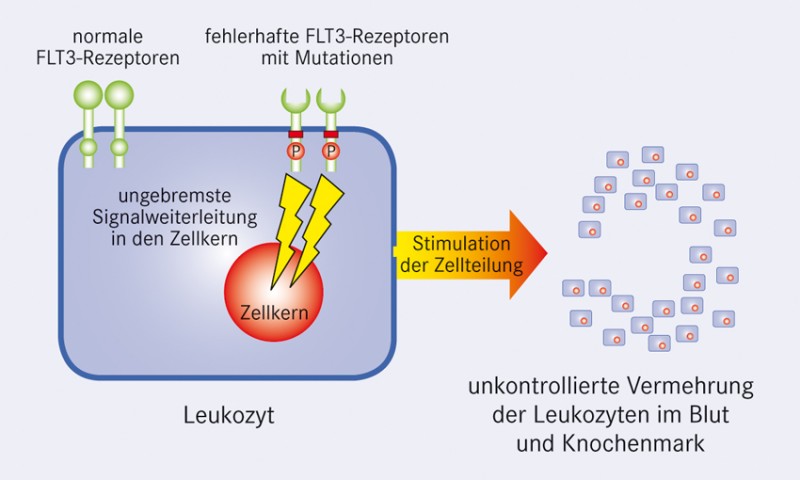
Mutationen der Rezeptor-Tyrosinkinase FLT-3
So konnten bei der akuten myeloischen Leukämie Mutationen eines Enzyms nachgewiesen werden, das die Zelldifferenzierung, das Zellwachstum und die Zellwanderung unreifer weißer Blutkörperchen reguliert, die Rezeptor-Tyrosinkinase FLT-3. Mit maßgeschneiderten kleinen Molekülen sollen die veränderten Rezeptor-Tyrosinkinasen blockiert und damit der Blutkrebs gestoppt werden. Erstmals ist das vor einem Jahrzehnt bei einer anderen Blutkrebsart, der chronisch myeloischen Leukämie gelungen. Mit dieser neuen Therapie konnte ein Langzeitüberleben der Patienten von über 90% erreicht werden. Auf ähnlich gute Erfolge hoffen die Forscher auch bei der AML. Sie behandelten in einer internationalen klinischen Phase IIb-Studie 95 AML-Patienten mit dem neuen Wirkstoff Midostaurin (N-Benzoylstaurosporin, PKC 412), einem Multitarget-Tyrosinkinase-Inhibitor, der das Zellwachstum mutierter Leukozyten verhindern soll. Bei etwa zwei Drittel der Patienten reduzierte sich die Zahl der unreifen Vorstufen der weißen Blutzellen um die Hälfte, damit konnte die Zahl leukämischer Zellen im Blut signifikant gesenkt werden. Bei einigen Patienten wurde auch der Krankheitsfortschritt zeitweilig gestoppt. Jetzt wird in einer Phase III-Studie untersucht, ob durch die Kombination mit einer Chemotherapie noch bessere Ergebnisse zu erreichen sind.
Quelle Fischer, T., et al.: J. Clin. Oncol. (2010) Vorabveröffentlichung
DOI: 10.1200/JCO.2009.27.6295.
hel



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.