














- DAZ.online
- DAZ / AZ
- DAZ 30/2014
- CYP2C und CYP2E1
Klinische Pharmazie
CYP2C und CYP2E1
Wichtige Enzyme für Arzneimittelinteraktionen
CYP2C-Isoenzyme
Alle CYP-Enzyme können gehemmt werden, dagegen ist eine ausgeprägte Induzierbarkeit nur für CYP1A2 und CYP3A4 beschrieben. Die strukturell sehr ähnlichen Isoenzyme CYP2C8, CYP2C9 und CYP2C19 machen ca. 18% der hepatischen CYP-Enzyme aus und sind am Stoffwechsel von ca. 10% aller hepatisch eliminierten Pharmaka beteiligt. CYP2C9 und CYP2C19 werden zudem auch im Gastrointestinaltrakt exprimiert (Tab. 1).
Hinsichtlich der pharmakogenetischen Varianten der CYP2C-Isoenzym-Aktivitäten sind die meisten „Kaukasier“ (Weiße) normale Metabolisierer (Tab. 2). Bei CYP2C19 erlaubt die pharmakogenetische Diagnostik in über 90% der Fälle eine Voraussage über die Enzymaktivität.
CYP2C9
CYP2C9 metabolisiert u.a. die endogenen Derivate der Arachidonsäure. Zu den wichtigsten arzneilichen Substraten von CYP2C9 gehören
- COX-hemmende Analgetika wie Diclofenac, Ibuprofen und Celecoxib,
- insulinotrope Antidiabetika wie Glibenclamid und Glimepirid sowie
- Vitamin-K-Antagonisten: die 4-Hydroxy-cumarin-Derivate Phenprocoumon und Warfarin (Tab. 3).
| Tab. 3: Substrate, Inhibitoren und Induktoren von CYP2C-Isoenzymen. Erläuterungen: * Elimination ist besonders stark von diesem Isoenzym abhängig; ! sehr geringe therapeutische Breite; + bis +++ Stärke der Inhibition oder Induktion | ||
| Substrate | Inhibitoren | Induktoren |
| CYP2C8 | ||
Montelukast *, Paclitaxel *!, Repaglinid * (anteilig weitere Substrate, siehe bei CYP2C9) | Gemfibrozil +++ Trimethoprim + | CAR- und PXR-Agonisten1 wie Barbiturate, Carbamazepin, Rifampicin Ritonavir + |
| CYP2C9 | ||
Insulinotrope Antidiabetika: Glibenclamid (aktiver Metabolit) ! Glimepirid (aktiver Metabolit) !, Nateglinid COX-Inhibitoren (NSAR): Celecoxib *, Diclofenac, Ibuprofen, Piroxicam wenig: Etoricoxib Antikonvulsiva: Phenytoin ! Sartane: Candesartan, Irbesartan, Losartan Antikoagulanzien: Warfarin *!, Phenprocoumon ! (teilweise, primär CYP3A4) Sonstige: Fluvastatin, Montelukast, Valproat | Amiodaron ++ Azol-Antimykotika: Fluconazol ++, Voriconazol Valproat Co-trimoxazol 5-Fluorouracil Fluoxetin Leflunomid Grapefruit (Naringin) + | CAR-, PXR- und GR-Agonisten1 wie Barbiturate, Carbamazepin, Rifampicin Ritonavir + |
| CYP2C19 | ||
PPI: Lansoprazol *, Omeprazol *, Pantoprazol *, Rabeprazol Antikonvulsiva: Diazepam *, Phenobarbital !, Phenytoin ! nicht: Lorazepam Antidepressiva: Amitriptylin * (auch CYP1A2 und CYP2D6), Citalopram * (auch CYP3A4 und CYP2D6), Clomipramin (teilweise), Fluoxetin (auch CYP2D6), Moclobemid * Sonstige: Clopidogrel (Prodrug), wenig: Prasugrel (Prodrug) Cyclophoshamid, Pentamidin *, Nelfinavir *, Thalidomid * | SSRI: Fluvoxamin +++, Fluoxetin +++ nicht: Citalopram Azol-Antimykotika: Fluconazol +++, Voriconazol PPI: Omeprazol ++, Esomeprazol ++ Ticlopidin +++, Topiramat + Grapefruit | CAR-, PXR- und GR-Agonisten1 wie Barbiturate, Carbamazepin, Rifampicin Ritonavir ++ Johanniskraut |
| 1 CAR = konstitutiver Androstanrezeptor; PXR = Pregnan-X-Rezeptor; GR = Glucocorticoidrezeptor (s. DAZ 2012, Nr. 40, S. 65) | ||
Durch CYP2C9-Inhibitoren oder bei langsamen CYP2C9-Metabolisierern kann es insbesondere bei den Antidiabetika und den Vitamin-K-Antagonisten – deren geringe therapeutische Breite bekannt ist – zu einer verstärkten Arzneimittelwirkung mit gravierenden Folgen wie Hypoglykämie bzw. Blutungen kommen. Bei den Vitamin-K-Antagonisten wird in den Fachinformationen und der ABDA-Datenbank vor einer Wirkungsverstärkung durch Amiodaron, Co-trimoxazol, Fluconazol, 5-Fluorouracil, Leflunomid, Metronidazol und Valproinsäure gewarnt (Tab. 3). Eine gleichzeitige Gabe sollte vermieden werden, zumindest sollte die Blutgerinnung engmaschig überprüft werden.
Beratungshinweis
Um mögliche Interaktionen von (Es-)Omeprazol mit CYP2C19-Substraten wie Clopidogrel zu vermeiden, empfiehlt sich der Wechsel auf Pantoprazol.
Auch die Anwendung von Glibenclamid und Glimepirid, die hauptsächlich über CYP2C9 metabolisiert werden, ist beratungsbedürftig, denn bei gleichzeitiger systemischer Anwendung von Azol-Antimykotika wie Fluconazol, Miconazol (Mundgel) und Voriconazol, die potente CYP2C9-Hemmstoffe sind, können hypoglykämische Symptome auftreten. Es ist wichtig, die Patienten darauf hinzuweisen und ihnen eine engmaschige Blutzuckerkontrolle zu empfehlen. Bei dermaler Applikation der Azol-Antimykotika treten diese Interaktionen nicht auf. Weitere relevante CYP2C9-Inhibitoren, die bei der Therapie mit Glibenclamid und Glimepirid das Risiko für Hypoglykämien steigern können, sind Co-trimoxazol und Fluoxetin.
Wenn ein Patient mit dem Antikonvulsivum Phenytoin – ebenfalls ein hauptsächliches Substrat von CYP2C9 – behandelt wird, sollte die Gabe von Co-trimoxazol nur unter Vorsicht in Erwägung gezogen werden, denn es wurden Fälle einer Phenytoin-Intoxikation berichtet. Ebenso sollte eine gleichzeitige Gabe von CYP2C9-Induktoren (s.u.) vermieden werden.
Fast alle COX-Inhibitoren (NSAR) werden teilweise über CYP2C9 metabolisiert. Hinweise auf klinisch relevante Wechselwirkungen liegen bisher nicht vor. Dennoch wird in den Fachinformationen von Celecoxib eine Dosisreduktion bei gleichzeitiger Gabe des CYP2C9-Inhibitors Fluconazol empfohlen sowie auf reduzierte Plasmaspiegel bei gleichzeitiger Anwendung von CYP2C9-Induktoren wie Rifampicin, Carbamazepin oder Barbituraten hingewiesen.
CYP2C19
Zu den wichtigsten CYP2C19-Substraten zählen Protonenpumpenhemmer (PPI), die teilweise zugleich CYP2C19 inhibieren, verschiedene Psychopharmaka und Clopidogrel (Tab. 3). Im Apothekenalltag sollte ein besonderes Augenmerk auf die PPI gelegt werden, insbesondere auf Omeprazol und Pantoprazol, die zeitlich limitiert und in niedriger Dosierung zur Selbstmedikation zugelassen sind.
Alle PPI werden schnell hepatisch metabolisiert, zuerst via CYP2C19 und im weiteren Verlauf über CYP3A4. Bei Patienten, die CYP2C19 langsam metabolisieren, sind aufgrund der guten Verträglichkeit dieser Substanzen keine toxischen Effekte zu erwarten. Dagegen kann ein schneller Abbau (bei schnellen Metabolisierern) die erwünschte Säurehemmung verhindern. In-vitro-Daten belegen für alle zugelassenen PPI, dass sie CYP2C19 konzentrationsabhängig hemmen. Eine klinische Relevanz lässt sich daraus aber nur für Omeprazol und sein Enantiomer Esomeprazol ableiten, da nach neuesten Studien einer ihrer Metaboliten CYP2C19 irreversibel hemmt [6]. Betroffen sind z.B. Patienten, die den Thrombozytenaggregationshemmer Clopidogrel und (zum Schutz vor gastrointestinalen Blutungen) zusätzlich ein PPI einnehmen. Clopidogrel ist ein Prodrug, das durch CYP2C19 aktiviert wird und bei einer CYP2C19-Hemmung unwirksam bleibt. In einem Positionspapier warnen die Deutschen Gesellschaften für Kardiologie (DGK) und für Verdauungs- und Stoffwechselkrankheiten (DGVS) vor der gleichzeitigen Anwendung von Omeprazol bzw. Esomeprazol und Clopidogrel. Da die CYP2C19-Hemmung irreversibel ist, ist auch die bisher empfohlene zeitlich versetzte Einnahme als nicht sinnvoll zu erachten.
Da die meisten beschriebenen Interaktionen von Omeprazol erst bei höheren Dosen und längerer Anwendungsdauer auftraten, ist seine Anwendung in der Selbstmedikation, die auf höchsten zwei bis vier Wochen begrenzt ist, als unkritisch zu beurteilen. Im Zweifelsfall sollte bei Risikopatienten Pantoprazol als Alternative empfohlen werden.
Auch Diazepam wird wesentlich durch CYP2C19 verstoffwechselt. Klinisch relevante Interaktionen wurden allerdings extrem selten berichtet, wahrscheinlich weil Diazepam auch durch CYP3A4 metabolisiert wird. Im Fall einer unvermeidbaren längerfristigen simultanen Einnahme mit Omeprazol wären Lorazepam oder Oxazepam als Alternativen vorzuziehen.
CYP2C8
CYP2C8 stellt mit gerade 7% nur einen geringen Anteil der CYP-Enzyme in der Leber und ist zudem nur zu 5% am Metabolismus der Arzneistoffe beteiligt, oft nur als sekundärer Stoffwechselweg. Für den Apothekenalltag relevante CYP2C8-Substrate sind Repaglinid und Paclitaxel, relevante CYP2C8-Inhibitoren sind Gemfibrozil und Trimethoprim (Tab. 3).
Klinisch wichtige Interaktionen sind für Repaglinid beschrieben. Die Kombination mit dem Fibrat Gemfibrozil, einem starken CYP2C8-Inhibitor, ist aufgrund des hohen Risikos für Hypoglykämien kontraindiziert. Als Alternativen bieten sich andere Fibrate wie Fenofibrat oder Bezafibrat an bzw. der Wechsel von Repaglinid zu Nateglinid. Für diese Kombinationen sind keine pharmakokinetischen Wechselwirkungen bekannt.
Ebenso besteht bei der gleichzeitigen Gabe von Repaglinid und Co-trimoxazol (Kombination von Trimethoprim und Sulfamethoxazol) ein erhöhtes Hypoglykämierisiko, wofür wahrscheinlich sowohl das Trimethoprim (CYP2C8-Hemmung) als auch das Sulfamethoxazol (Sulfonylharnstoff-ähnliche Wirkung) verantwortlich sind. Bei gleichzeitiger Anwendung von Repaglinid und schwachen CYP2C8-Induktoren wie Johanniskraut, Carbamazepin oder Rifampicin sollte Repaglinid gegebenenfalls höher dosiert werden.
In der Fachinformation von Paclitaxel ist die CYP2C8- vermittelte Metabolisierung als wichtiger Stoffwechselweg beschrieben. Daher wird von der gleichzeitigen Verabreichung von CYP2C8-Hemmern oder -Induktoren abgeraten.
CYP-Enzyme, Lakritze und Cortison
Cytochrom-P450-Enzyme sind nicht nur für die Pharmakokinetik von Arzneistoffen interessant, sondern können auch Zielstruktur von Arzneistoffen sein.
CYP51 dient Pilzen zur Sterolsynthese für die Zellmembran. Azol-Antimykotika (z.B. Itraconazol, Ketoconazol) blockieren diesen Syntheseweg, indem der Stickstoff des Imidazolrings die Hämgruppe des CYP51 blockiert, und wirken daher antimykotisch.
Das Antikonvulsivum Phenytoin und das Injektionsnarkotikum Etomidat sind ebenfalls Imidazolderivate und blockieren CYP11B1, auch bekannt als 11-beta-Hydroxylyase, das für den Steroidmetabolismus essenziell ist. Unter Phenytoin-Dauertherapie kommt es zu Virilisierungserscheinungen bei Frauen, und unter längerer Etomidat-Infusion sinkt die Cortisolkonzentration. Etomidat, Ketoconazol, Metyrapon (in Deutschland nicht zugelassen, nur Einzelimport nach § 73 AMG) werden off-label bei vital bedrohlichen Hypercortisolämien (z.B. präoperativ bei M.Cushing) eingesetzt.
Glycyrrhizin, ein Inhaltstoff der Lakritze, hemmt CYP11B2 und CYP17A1 sowie die 11-beta-Hydroxysteroid-Dehydrogenase (11β-HSD, ein Nicht-Cytochrom-P450-Enzym). Ohne CYP11B2- und 11β-HSD-Aktivitäten steigt die Synthese von Mineralocorticoiden, die an der Niere eine Wasserretention mit nachfolgender Hypertonie verursachen. CYP17A1 katalysiert die Umwandlung von weiblichen in männliche Geschlechtshormone. Starklakritze in hohen Dosen kann daher die Testosteronkonzentration senken (klinisch nicht relevant).
Die CYP17A1-Inhibition durch Abirateron senkt die Plasmakonzentrationen männlicher Geschlechtshormone, Abirateron wird deshalb bei Patienten mit Prostatakarzinom angewendet.
CYP2E1
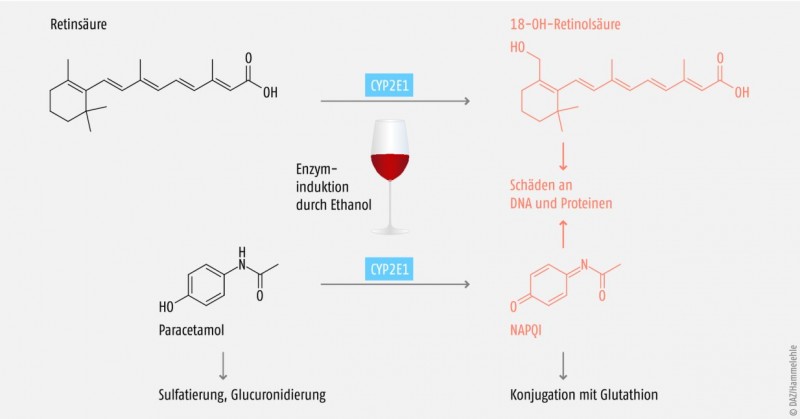
CYP2E1 macht ca. 7% der CYP-Enzyme in der Leber aus und ist an der Elimination von ca. 2% der hepatisch eliminierten Pharmaka beteiligt. Darunter finden sich vor allem Inhalationsnarkotika wie Sevofluran, weshalb eine CYP2E1- Induktion durch Alkoholabusus oder Isoniazid insbesondere vor einer Allgemeinanästhesie mit Fluranen abgeklärt werden muss (Tab. 4).
Regelmäßiger Alkoholkonsum (30–60 g/d bei Männern, 20–40 g/d bei Frauen; 60 g entspricht 0,7 l Wein oder 1,5 l Bier) steigert die Aktivität von CYP2E1, da Ethanol einerseits das CYP2E1 vor dem Abbau schützt und andererseits die Biosynthese von CYP2E1 steigern kann [10, 11].
Paracetamol wird, wenn auch nur zu einem geringen Teil, ebenfalls über CYP2E1 zum lebertoxischen Metaboliten N‑Acetyl-4-benzochinonimin (NAPQI) umgewandelt. Bei chronischem Alkoholabusus kommt es über eine CYP2E1-Induktion zur vermehrten Bildung dieses lebertoxischen Metaboliten, und somit steigt das Risiko für Leberzellnekrosen, hepatozelluläre Insuffizienz und Koma (Abb. 1). Retinsäure (Vitamin-A-Säure) wird auch über CYP2E1 abgebaut (Abb. 1). Demzufolge kann es bei Alkoholikern aufgrund des beschleunigten hepatischen Metabolismus zu Symptomen eines Vitamin-A-Mangels kommen (z.B. erhöhte Infektanfälligkeit, Eisenmangel, Müdigkeit) [14]. Da aber einige Zwischenprodukte des Retinsäureabbaus apoptotisch wirken oder die Hyperproliferation und Kanzerogenese in verschiedenen Geweben fördern können, sollten Alkoholiker eher kein Vitamin A supplementieren. Es wird empfohlen, den erhöhten Bedarf durch den Verzehr carotinhaltiger Lebensmittel zu decken [12].
Diese Artikelversion wurde am 5.9.14 korrigiert.
Literatur
[1] Shin HC, et al. Comparative gene expression of intestinal metabolizing enzymes. Biopharm Drug Dispos 2009;30(8):411–421
[2] Cooke MJ, Waring WS. Citalopram and cardiac toxicity. Eur J Clin Pharmacol 2013;69(4):755-760
[3] Rodenburg EM, et al. Genetic variance in CYP2C8 and increased risk of myocardial infarction. Pharmacogenet Genomics 2010;20(7):426-434
[4] Mrazek DA, et al. CYP2C19 variation and citalopram response. Pharmacogenet Genomics 2011;21(1):1-9
[5] Yang LJ, et al. Effects of allicin on CYP2C19 and CYP3A4 activity in healthy volunteers with different CYP2C19 genotypes. Eur J Clin Pharmacol 2009;65(6):601-608
[6] Lu FM, et al. [Impact of proton pump inhibitor omeprazole on the antiplatelet effect of clopidogrel in individuals with various CYP2C19*2 genotypes] (auf Chinesisch). Chin J Med Genet 2012;29(4):478-481
[7] Uno T, et al. Lack of significant effect of grapefruit juice on the pharmacokinetics of lansoprazole and its metabolites in subjects with different CYP2C19 genotypes. J Clin Pharmacol 2005;45(6):690-694
[8] Lilja JJ, et al. Effects of clarithromycin and grapefruit juice on the pharmacokinetics of glibenclamide. Br J Clin Pharmacol 2007;63(6): 732-740
[9] Trenk D, et al. Personalizing antiplatelet therapy with clopidogrel. Clin Pharmacol Ther 2012;92(4):476-485
[10] Roberts BJ, et al. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J Biol Chem 1995;270(50):29632-5
[11] Zhong Y, et al. Induction of brain CYP2E1 by chronic ethanol treatment and related oxidative stress in hippocampus, cerebellum, and brainstem. Toxicology 2012;302(2–3):275-284
[12] Biesalski HK, Fürst P, Kasper H. Ernährungsmedizin, 4. Aufl. Thieme, Stuttgart 2010
[13] Preda VA, et al. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol 2012;167:137-143
[14] Semba RD. The role of vitamin A and related retinoids in immune function. Nutr Rev 1998;5:S38-48
Autoren
Dr. rer. nat. Kirstin Reinecke, Dr. med. Ruwen Böhm, Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi, Prof. Dr. med. Thomas Herdegen
Institut für Experimentelle und Klinische Pharmakologie
UK-SH, Campus Kiel
Hospitalstr. 4, 24105 Kiel
Rückblick
Von denselben Autoren sind folgende Beiträge über CYP-Enzyme in der DAZ erschienen:
- Arzneimittelinteraktionen verstehen, vermitteln und vermeiden.
DAZ 2012, Nr. 36, S. 64–74 - Interaktionen mit CYP3A4.
DAZ 2012, Nr. 40, S. 58–67 - Arzneimittel und CYP2D6.
DAZ 2012, Nr. 47, S. 60–66 - Arzneimittel und CYP1A2.
DAZ 2013, Nr. 22, S. 44–49



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.