












- DAZ.online
- News
- Spektrum
- Die FDA fand schon 2017 ...
Zhejiang Huahai Pharmaceutical
Die FDA fand schon 2017 eine unbekannte Verunreinigung beim Valsartan-Hersteller
Stuttgart - 07.09.2018, 07:00 Uhr
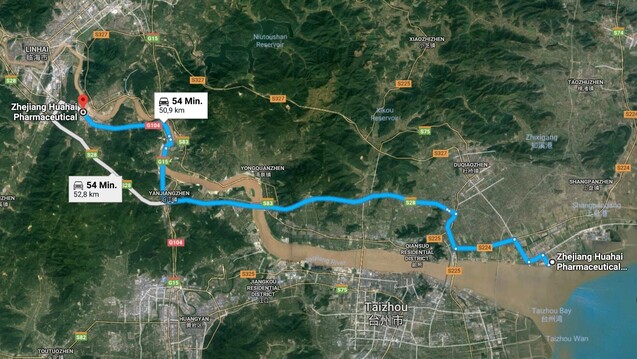
Zhejian Huahai Pharmaceutical besitzt zwei Herstellungsstandorte in direkter Nähe zueinander. (Foto: Screenshot / Google Maps)
Die Ergebnisse der FDA-Inspektionen beim Valsartan-Hersteller
Die Eingabe des Namens des Valsartan-Herstellers in der FDA-Datenbank reicht und schon werden die Inspektionen der vergangenen Jahre bei Zhejiang Huahai Pharmaceutical aufgelistet (nicht klassifizierte Inspektionen erscheinen nicht) – mit zwei unterschiedlichen Addressangaben: Taizhou und Linhai.
Die Addresse, die im Zusammenhang mit dem viel diskutierten CEP-Zertifikat auftaucht, beinhaltet die Stadt Linhai (Postleitzhal: 317024). Dort wurde der Wirkstoffhersteller laut der FDA-Datenbank 2010, 2014 und 2017 inspiziert. Während 2010 und 2014 noch keine Maßnahmen erforderlich schienen (NAI), wurden nach der Inspektion im Mai 2017 freiwillige Maßnahmen des Herstellers als notwendig erachtet. Auch am Standort Taizhou schien es bei einer Inspektion 2016 Unstimmigkeiten zu geben.
Gab es auch europäische Inspektionen?
Sucht man in der EudraGMDP-Datenbank nach Inspektionsdaten in China mit dem Standort unter der Postleitzahl 317024, ist erkennbar, dass zuletzt im März 2018 eine europäische Inspektion bei Zhejiang Huahai Pharmaceutical stattgefunden hat. Mängel wurden aber scheinbar keine festgestellt, denn „Non-Compliance-Reports“ sind in der Liste nicht aufgeführt.
Ein Blick auf die älteren Inspektionsdaten verrät, dass auch eine deutsche Behörde Zhejiang Hauhai Phamaceutical in China häufiger besucht hat: „Freie und Hansestadt Hamburg, Ministry of Health and Consumer Protection“ liest man auf dort hinterlegten Dokumenten.
Was wurde von der FDA bemängelt? Das Formular 483
Wenn die FDA bei einer Inspektion Mängel entdeckt, werden diese in sogenannten 483-Formularen festgehalten. Diese Formulare sind bei ausländischen Inspektionen jedoch nicht einfach über eine Datenbank zugänglich.
Schon im August berichtete die Zeitung „Die Welt“ über „Geister-Werte“ im Fall Valsartan und bezog sich dabei auf „zwei Prüfberichte über die chinesische Firma Zhejiang Huahai“, die von der FDA veröffentlicht worden seien. Die Protokolle aus den Jahren 2016 und 2017 belegen laut Welt „deutlich, dass bei dem Hersteller in der ostchinesischen Provinz Zhejiang südlich von Shanghai offenkundig einiges im Argen liegt“. Über den Internetauftritt der FDA konnte DAZ.online besagte Dokumente nicht auffinden. Eine Google-Suche hilft aber weiter und leitet direkt zu zwei PDF-Dokumenten, die von der FDA zu stammen scheinen. In beiden Fällen handelt es sich um das Formular 483.
Welche Mängel von der FDA festgehalten wurden
Das Formular 483, das sich auf eine Inspektion vom 14. bis
18. November 2016 bezieht, richtet sich nicht nur namentlich an den Valsartan-Wirkstoffhersteller
Zhejiang Huahai Pharmaceutical Co., sondern ist auch an dieselbe
Adresse adressiert, unter der das CEP beim EDQM hinterlegt ist. Dabei handelt es sich nicht um einen reinen Produktionsstandort für
Wirkstoffe, sondern auch für Fertigarzneimittel (auf Karte weiter im Inland
gelegener Standort, siehe Screenshot oben). Welchen Herstellungsprozess die Mängel genau betreffen,
wurde geschwärzt. Allerdings sind einige Beobachtungen aufgeführt:
1. Beobachtung
„Schriftliche Anweisungen, die dazu dienen sollen, sterile Produkte vor einer Verunreinigung zu schützen, werden nicht befolgt.“ Dieser Vorwurf wird mit konkreten Beobachtungen begründet, die aufgrund von Schwärzungen seitens der FDA nicht sinnhaft wiedergegeben werden können.
2. Beobachtung
„Es werden keine Laborkontrollen durchgeführt, die auf wissenschaftlich fundierten und angemessenen Spezifikationen, Standards, Probenplänen und Prüfverfahren basieren und die dafür entworfen wurden, Identität, Stärke, Qualität und Reinheit der Produkte sicherzustellen.“ Auch dieser Kritikpunkt scheint vor allem die Reinraumbedingungen zu betreffen. So würden Umgebungsmessungen bei geöffneten Türen schlichtweg nicht stattfinden. Zudem werde eine etwaige Endotoxin-Belastung nicht evaluiert.
3. Beobachtung
„Reinigung und Desinfektion der Geräte weisen Mängel auf.“ So sollen beispielsweise Rohrleitungen nicht regelmäßig inspiziert und gereinigt werden. Am Tag der Inspektion wurde in einer Rohrleitung weißer Staub auf der Oberfläche entdeckt, der nicht näher identifiziert wurde.
4. Beobachtung
„Daten werden nicht zeitgleich dokumentiert.“ Die „checked by“-Einträge bestimmter Tabletten-Chargen würden nicht zum Zeitpunkt der Chargen-Produktion dokumentiert. Der für die Umweltüberwachung verantwortliche QC-Analytiker soll zudem die Stichprobenziehung nicht zum Zeitpunkt ihres Auftretens dokumentieren.



2 Kommentare
Die FDA fand schon 2017 eine unbekannte Verunreinigung beim Valsartan-Hersteller
von Dr. Christoph Sonntag am 07.09.2018 um 18:08 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Die FDA fand schon 2017 eine unbekannte Verunreinigung beim Valsartan-Hersteller
von Dr. Christoph Sonntag am 07.09.2018 um 17:59 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Das Kommentieren ist aktuell nicht möglich.