











- DAZ.online
- News
- Pharmazie
- Glitazare und GLP-1 – ...
Gescheiterte PPAR-Agonisten
Glitazare und GLP-1 – neuer Ansatz bei Diabetes?
Düsseldorf - 19.09.2022, 07:00 Uhr
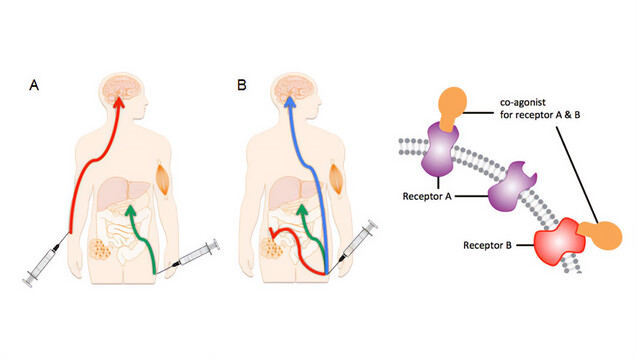
Forschern des Helmholtz Zentrums München ist es gelungen, die blutzuckersenkenden Effekte von GLP-1 und Tesaglitazar in einem einzigen hochwirksamen Molekül zu vereinen (B). (b/Bild: helmholtz-munich.de)
PPAR-Agonisten sind auf der einen Seite vielversprechende Wirkstoffe, die für einen Einsatz gegen viele „Zivilisationskrankheiten“ wie Diabetes oder nicht-alkoholische Fettleber erforscht werden. Die Klasse der Glitazare allerdings, die zwei Arten von PPA-Rezeptoren aktivieren, galt spätestens seit 2013 als gescheitert, da sie ungeahnte unerwünschte Wirkungen zeigte. Nun haben Forscher:innen des Helmholtz Zentrums München gemeinsam mit Wissenschaftler:innen des Deutschen Zentrums für Diabetesforschung (DZD) und Novo Nordisk einen neuen Ansatz dafür aufgezeigt.
PPAR, das steht für Peroxisom-Proliferator-aktivierte Rezeptoren, von denen drei als Alpha, Beta/Delta und Gamma bezeichnete Subtypen bekannt sind. Diese innerhalb der Zelle verorteten Rezeptoren steuern nach ihrer Aktivierung durch einen Liganden als Transkriptionsfaktoren die Aktivierung einer ganzen Reihe von Genen, die mit dem Stoffwechsel in Verbindung stehen.
- Wird Alpha aktiviert, sinken etwa die Blutfettwerte und Entzündungsprozesse werden gehemmt.
- Beta/Delta, der oft nur als Delta bezeichnet wird, reguliert ebenfalls den Fettstoffwechsel und zeigt im Tierversuch an adipösen Mäusen, dass seine Aktivierung unter anderem das Körpergewicht senkt.
- Gamma schließlich spielt eine wichtige Rolle im Glucosestoffwechsel und bei der Insulinsensitivität.
Mehr zum Thema

Wenn die Leber ohne Bier und Korn ihr Fett abkriegt
Herausforderung nichtalkoholische Fettleber

Lanifibranor überzeugt bei nichtalkoholischer Steatohepatitis
Neuer Arzneistoff gegen Fettleber

Die hepatische Manifestation des metabolischen Syndroms
Nichtalkoholische Fettlebererkrankung
Damit sind diese Rezeptoren, die nahezu in allen Körperzellen exprimiert werden, spannend für die pharmazeutische Forschung – und etwa mit den Wirkstoffklassen der Fibrate (beispielsweise Bezafibrat, Gemfibrozil) als Lipidsenker (als Agonisten von PPAR Alpha) oder der Glitazone (beispielsweise Pioglitazon) als „Insulin-Sensitizer“ (Agonisten von PPAR Gamma) bereits im therapeutischen Einsatz.
Duale PPAR-Agonisten bislang gescheitert – Pan-Agonist dagegen nicht
Der Zusammenhang von Blutzucker und Blutfettwerten bei Krankheiten wie Typ-2-Diabetes brachte Forscher:innen in der Vergangenheit auf die naheliegende Idee, Wirkstoffe zu finden, die als duale PPAR-Agonisten wirken, gegen zwei der Rezeptoren – oder auch gegen alle drei als „Pan-PPAR-Agonisten“. Wie kompliziert und oft noch unverstanden allerdings das biochemische Zusammenspiel im Organismus ist, lässt sich in dem Zusammenhang daran zeigen, dass die Substanzklasse der Glitazare als duale PPAR-Agonisten, die Alpha und Gamma aktivieren, seit spätestens 2013 als gescheitert galt, während gerade erst der Pan-PPAR-Agonist Lanifibranor auf dem Weg zur schnellen Zulassung in den USA ist.
Eine Übersicht über das Potenzial neuer Wirkstoffe und Therapieregime
Therapie des Typ-2-Diabetes im Wandel
Vertreter der Glitzare sind Tesaglitazar, Muraglitazar, Ragaglitazar und zuletzt Aleglitazar. Tesaglitazar galt bis 2006 als ein Hoffnungsträger, der die Insulinempfindlichkeit erhöhen und gleichzeitig die Blutfettwerte senken sollte. Im Mai 2006 stellte AstraZeneca jedoch die Forschung in der Phase III ein, da sich nicht nur Nierenprobleme, sondern auch eine Cardiotoxizität gezeigt hatte. Muraglitazar und Ragaglitazar scheiterten bereits etwas früher und wurden ebenfalls mit kardiotoxischen und kanzerogenen Wirkungen in Zusammenhang gebracht. Zuletzt stoppte das Unternehmen Roche die Erforschung eines angepassten Wirkstoffes namens Aleglitazar im Jahr 2013 wegen ähnlicher unerwünschter Wirkungen – und musste dafür viel Kritik einstecken und viel Geld abschreiben.
Nun allerdings könnten die Multi-PPAR-Agonisten nicht nur wegen Lanifibranor ein Revival erleben. Forscher:innen des Helmholtz Zentrums München gemeinsam mit Wissenschaftler:innen des Deutschen Zentrums für Diabetesforschung (DZD) und Novo Nordisk haben inzwischen einen neuen Ansatz präsentiert, mit dem sie die als gescheitert geltende Wirkstoffklasse der Glitazare neu beleben könnten. Ihre Ergebnisse veröffentlichten sie jetzt im Fachmagazin Nature Metabolism.
Kopplung an GLP-1 vermittelt den Wirkstoff zielgerichtet
Der vergleichsweise einfache Ansatz dabei: Sie koppelten das zur Klasse der „Small Molecules“ gehörende Tesaglitazar an das Peptidhormon GLP-1 (Glucagon-like Peptide 1). Dieses „Darmhormon“ wirkt sehr spezifisch im Magen und Darm und „adressiert“ daher das daran gekoppelte Tesaglitazar auch nur an Zellen und Gewebe, die den Rezeptor für GLP-1 tragen. GLP-1 beziehungsweise analoge Mimetika sind als blutzuckersenkende Arzneimittel im Einsatz.
„Durch diesen Kniff ist es uns gelungen, die blutzuckersenkenden Effekte von GLP-1 und Tesaglitazar in einem einzigen hochwirksamen Molekül zu vereinen und gleichzeitig Tesaglitazar aus Geweben fernzuhalten, in denen es schädliche Effekte hat“, erklärt Studienautor PD Dr. Timo Müller, Direktor des Instituts für Diabetes und Adipositas am Helmholtz Zentrum München und Leiter der Abteilung für Molekulare Pharmakologie.
Im Tierversuch mit Mäusen erzielten die Forscher mit dem kombinierten Wirkstoff vielversprechende Ergebnisse – wohl ohne die gefürchteten Wirkungen auf Herz und Niere. „In männlichen adipösen und diabetischen Mäusen verbessert er den Zuckerstoffwechsel deutlich stärker als die alleinige Behandlung mit den einzelnen Hormonen GLP-1 oder Tesaglitazar – und dies ohne schädliche Nebenwirkungen auf die Leber oder Niere“, sagt Studienautorin Dr. Kerstin Stemmer, Professorin für Molekulare Zellbiologie an der Universität Augsburg. Besonders effektiv habe der Wirkstoff die Glukosetoleranz gesteigert. Es reichten minimale Dosierungen des neuen Wirkstoffs, um den Blutzuckerstoffwechsel nachhaltig zu verbessern, sagt Stemmer.
Optimistisch, unerwünschte Wirkungen minimieren zu können
Müller zeigt sich optimistisch, dass sich auch am Menschen so die unerwünschten Effekte reduzieren lassen. „Kein Experiment der Maus ist in der Lage, kardiotoxische Effekte im Menschen gänzlich auszuschließen. Mäuse sind sehr insensitiv gegenüber CV-Effekten. Jedoch wurde Tesaglitazar (bei frühere Forschungen) in der Regel oral mit einer etwa 1000-fach höheren Dosierung gegeben als unser peripher appliziertes GLP-1/Tesaglitazar, was alleine schon stark darauf hindeutet, dass weniger Nebeneffekte zu erwarten sind. Dazu kommt, dass GLP-1/Tesaglitazar nur in Zellen gelangt, welche den GLP-1 Rezeptor exprimieren. Und dieser ist nur in ganz wenigen Zellen im Herzen zu finden“, sagt er.
GLP-1-Rezeptoragonisten, DPP-4- und SGLT-2-Inhibitoren punkten unterschiedlich
Drei Antidiabetika-Klassen im Vergleich
Bei hohem kardiovaskulärem Risiko sind GLP-1-Rezeptoragonisten und SGLT-2-Hemmer erste Wahl
Paradigmenwechsel bei Diabetes
Auch Dulaglutid überzeugt mit protektiven Effekten
GLP-1-Rezeptoragonisten im Aufwind
In ihrer Arbeit betonen die Forscher auch, dass das konjugierte Wirkstoffmolekül nicht einfach nur ein verstärkter GLP-1-Ligand ist, sondern spezifisch auch an PPAR Gamma bindet.
Generell könne man die Ergebnisse wohl auch auf die anderen Vertreter der Glitazare übertragen. „Andere Glitazare haben konjugiert an GLP-1 sehr ähnliche Effekte. Tesaglitazar war nicht unser einziges Molekül. Da Tesaglitazar in den klinischen Studien etwas sicherer war als andere Glitazare, haben wir uns dann für die Publikation von GLP-1/Tesaglitazar entschieden“, sagt Müller.
Die Veränderung des Wirkstoffs hat allerdings auch Folgen für die Form der Verabreichung. So war Tesaglitazar vor allem für eine orale Aufnahme konzipiert worden. Die Forscher:innen der neuen Studie injizierten den Wirkstoff nun aber ihren Versuchstieren. „In allen Studien wurde GLP-1/Tesaglitazar peripher (subkutan) appliziert. Das Molekül ist, wie auch andere GLP-1-Mimetika, nicht resistent gegenüber der Hydrolyse durch die Magensäure“, erklärt Müller.
GLP-1-Kopplung bereits in der Vergangenheit mit anderen Wirkstoffen erfolgreich
Darüber hinaus wird GLP-1 im Körper in der Regel schnell durch ein Enzym namens DPP-4 (Dipeptidylpeptidase-4) abgebaut. Dem musste man mit einem weiteren biochemischen Trick vorbeugen. „Tesaglitazar ist ein sogenanntes small Molecule, kein Peptid wie GLP-1, und wurde an das C-terminale Ende von GLP-1 konjugiert. Das verwendete GLP-1 ist durch Modifikation der DPP-4 sensiblen 2’ten N-terminalen Aminosäure resistent gegenüber DPP-4.“ Dabei habe man an Position 2 des Peptids die Aminosäure Alanin durch Aminoisobuttersäure ersetzt. Die Resistenz gegenüber DPP-4 habe also nichts mit dem Tesaglitazar zu tun, sagt er – DDP-4 erkenne und spalte ein Dipeptid vom N-terminalen Ende des Peptids, während Tesaglitazar am anderen Ende (dem C-Terminus) konjugiert sei.
Mit der GLP-1-Kopplung habe man bereits in der Vergangenheit Erfolge erzielt. Beispielsweise mit GLP-1/Östrogen (Finan et al., Nature Medicine 2013 und Sachs et al., Nature Metabolism 2020), GLP-1/Dexamethason (Quarta et al., Cell Metabolism 2017) und Glucagon/T3 (Finan et al., Cell 2016).
Als nächste wolle man nun versuchen, das Molekül weiter biochemisch zu optimieren, sagt Müller. „Wir denken, dass der Effekt noch weiter verstärkt werden kann.“ Wie lange es jedoch dauern könnte, bis GLP-1/Tesaglitazar in die klinische Erprobung gehen könnte, sei unklar. „Diese Frage lässt sich schwer beantworten, da dies im Ermessen des Industriepartners liegt. Wann das Molekül in eine klinische Entwicklung geht, lässt sich derzeit nicht vorhersagen“, sagt der Wissenschaftler.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.