















- DAZ.online
- DAZ / AZ
- DAZ 38/2016
- Noch sicher wirksam oder ...

Foto: Narong Jongsirikul – Fotolia.com
Zytostatika-Ausschreibungen
Noch sicher wirksam oder schon verfallen?
Zytostatika-Zubereitungen und die Aussagekraft der Herstellerangaben für die Haltbarkeit
Dies sind die „Player“ auf dem „Spielfeld“ der Zytostatika-Ausschreibungen:
- Ärzte und Kliniken,
- bisher beliefernde Apotheken,
- neue beliefernde Apotheken und
- die Krankenkassen.
Ärzte und Kliniken möchten in der Regel die bestehenden Lieferstrukturen und Ansprechpartner erhalten bzw. haben weitere Anreize, einen neuen, von der Krankenkasse bestimmten, Lieferanten (Apotheke) abzulehnen.
Apotheken als bisheriger Lieferant haben ein nachvollziehbares Interesse, ihre teuren Investitionen z. B. für die Einrichtung des Reinraumes (nach ApBetrO 2012) zu amortisieren. Hier laufen auch noch andere Warenströme zusammen (im Sinne einer Teil- oder Komplettversorgung der Praxis / der Klinik).
Apotheken als neuer Lieferant möchten die Auslastung ihrer bestehenden Zytostatika-Herstellung erhöhen bzw. die anfallenden Fixkosten durch mehr Kunden und höhere Stückzahlen besser verteilen. Auch hier wird eine zusätzliche Teil- oder Komplettversorgung angestrebt. Apotheken ohne eigene Sterilherstellung erhoffen sich neue Kundenkontakte und Zusatzgeschäfte (und natürlich auch zusätzlichen Gewinn).
Den Krankenkassen geht es aktuell mit Sicherheit vor allem um Einsparungen und Optimierungen. In naher oder mittlerer Zukunft erhoffen sich die Kassen vermutlich bessere Verhandlungsmöglichkeiten mit den (wenigen) verbliebenen Anbietern (Herstellern). Ein umgekehrtes Szenario ist ebenso denkbar: Wenige noch verbliebene große Apotheken oder Herstellungsbetriebe nutzen ihre Marktdurchdringung bzw. die mangelnde Konkurrenz, um dann den Krankenkassen Preiserhöhungen zu diktieren.
Was das Arzneimittelgesetz sagt
Nach dem Arzneimittelgesetz (§ 11a Absatz 1 Nr. 6. c) AMG) ist der pharmazeutische Unternehmer verpflichtet, in der Fachinformation Angaben zur „Dauer der Haltbarkeit und, soweit erforderlich, die Haltbarkeit bei Herstellung einer gebrauchsfertigen Zubereitung des Arzneimittels oder bei erstmaliger Öffnung des Behältnisses“ bekannt zu machen. Hier geht es um die Angaben, welche momentan so sehr im Zentrum der Diskussion stehen. Der Arzneimittelrecht Kommentar (Kloesel/Cyran, Stand 2016, 131. Akt.-Lief.) bringt hierzu keine ergänzenden Informationen.
Auf der Primär- und Sekundärverpackung steht das Verfalldatum des Arzneimittels (AMG § 10 Absatz 1 Nr. 9.). Es ist Teil der Zulassung durch das BfArM und wird vom Hersteller auf der Grundlage von Untersuchungen zur Stabilität, den Lagerungsbedingungen und anderer Parameter festgelegt. Der Wirkstoffgehalt soll bis zum Ende der Laufzeit noch bei 90 Prozent liegen. Er kann aber auch noch 98 Prozent betragen (was ein viel späteres Verfalldatum zulassen würde).
Das BfArM prüft nicht den wissenschaftlichen Hintergrund des Verfalldatums. Es wird vom Hersteller unter Berücksichtigung verschiedener Gesichtspunkte festgelegt. Eine zu kurze Haltbarkeit kann im Falle von Herstellproblemen zu Defekten führen, eine zu lange Haltbarkeit wiederum führt zu weniger Umsatz oder seltenerem Austausch gegen frische Ware bei den zu bevorratenden Arzneimitteln.
Der Arzneimittelrecht Kommentar (Kloesel/Cyran, § 10 AMG, Punkt 59) stellt hierzu fest:
Eine Verlängerung des Verfalldatums eines im Verkehr befindlichen Arzneimittels ist nicht möglich. Dies kann den Tatbestand der Fälschung erfüllen (vgl. § 8). Die unternehmerische Haftung des pharmazeutischen Unternehmers entfällt. Wenn allerdings durch „Follow-up“-Stabilitätsuntersuchungen eine längere Haltbarkeit gewährleistet werden kann, ist dies der Zulassungsbehörde anzuzeigen. Anschließend können die Chargen mit einem längeren Verfalldatum gekennzeichnet werden.
Derselbe Kommentar zu § 10 AMG (und nicht, wie zu erwarten, zu § 11a AMG) behandelt unter Punkt 62a. das Thema „Aufbrauchfristen“:
Vom Verfalldatum sind die Aufbrauchfristen zu unterscheiden. Dem Verfalldatum liegen Haltbarkeitsversuche mit einem konkreten Arzneimittel in seinem geschlossenen Behältnis unter Einhaltung der vorgesehenen Lagerungsbedingungen im Handel zugrunde. […] Von Aufbrauchfristen wird gesprochen, wenn es um die Haltbarkeit des Arzneimittels im Behältnis (eines meist aseptisch oder steril zubereiteten Arzneimittels) nach Öffnung (Anbruch) – auch wiederholt – geht oder für den Gebrauch noch zubereitet werden muss. Angaben über die Haltbarkeit nach Öffnung des Behältnisses sind für den Anwender des Arzneimittels von Bedeutung und resultieren in der Angabe von empfohlenen Aufbrauchfristen in der Packungsbeilage […]. Die Verantwortung liegt in der Regel nicht beim pharmazeutischen Unternehmer, weil er dieses Handeln nicht beeinflussen kann. Jedoch muss der pharmazeutische Unternehmer Unterlagen über die Haltbarkeit nach Öffnung des Behältnisses zusammen mit dem Zulassungsantrag vorlegen.
Änderung des Verfalldatums – z. B. Tamiflu®
Das angegebene Verfalldatum wie auch die Haltbarkeit nach Anbruch (Aufbrauchfrist) können sich nachträglich ändern, sie sind nicht das „Maximum“ und auch nicht das „Minimum“. Dies zeigt sich sehr deutlich in § 71 Absatz 1 AMG:
Die in § 10 Abs. 1 Nr. 9 vorgeschriebene Angabe des Verfalldatums kann entfallen bei Arzneimitteln, die an die Bundeswehr, die Bundespolizei sowie für Zwecke des Zivil- und Katastrophenschutzes an Bund oder Länder abgegeben werden. Die zuständigen Bundesministerien oder, soweit Arzneimittel an Länder abgegeben werden, die zuständigen Behörden der Länder stellen sicher, dass Qualität, Wirksamkeit und Unbedenklichkeit auch bei solchen Arzneimitteln gewährleistet sind.
Die zuständigen Ministerien (Verteidigung, Inneres) legen die entsprechenden Untersuchungen fest, um die pharmazeutische Qualität, Wirksamkeit, Unbedenklichkeit der jeweiligen Arzneimittel zu gewährleisten.
Hierzu ein Beispiel: Während der weltweiten Influenza-Pandemie hat am 11. Mai 2009 die Europäische Kommission einer Verlängerung der Haltbarkeit der Tamiflu® Kapseln (30 mg, 45 mg und 75 mg Oseltamivir) von fünf auf sieben Jahre zugestimmt. Dementsprechend beträgt die Haltbarkeit der Kapseln nunmehr sieben Jahre. Gleichzeitig wurde der Lagerungshinweis „Nicht über 25 °C lagern“ in die entsprechenden Fach- und Gebrauchsinformationen aufgenommen sowie auch auf den Faltschachteln aktualisiert. Für Chargen, die vor der Änderung der Haltbarkeits- und Lagerungsvorschriften in Verkehr gebracht wurden, gelten weiterhin die jeweils vom Hersteller aufgedruckten Haltbarkeitsdaten (AMK 19.05.2009 und 14.07.2009).
Grund für die Verlängerung der Haltbarkeit war damals eine Verknappung des Wirkstoffes Oseltamivirphosphat infolge der Verknappung des Eduktes Shikimisäure.
Das SLEP-Programm in den USA
In den USA wurden in den 1980er-Jahren federführend durch das U.S. Department of Defense (DoD) unter Beteiligung der FDA und des U.S. Department of Health and Human Services (HHS) weitergehende Stabilitätsuntersuchungen bezüglich des Strategic National Stockpile (SNS) initiiert: das SLEP-Programm (Shelf-Life Extension Program) [1, 2]. Hintergrund waren ursprünglich die enormen Kosten für die Beseitigung der abgelaufenen Medikamente und die Neubeschaffung. Im Jahr 2005 berichtete das DoD, dass die Untersuchungen des SLEP-Programmes Kosten von 350.000 $ pro Jahr verursachten, die Einsparungen beliefen sich auf 33.000.000 $ – ein Verhältnis von 1 zu 94! Die Untersuchungen führten zu bemerkenswerten Ergebnissen: Unter optimalen Lagerungsbedingungen konnte beispielsweise die Haltbarkeit von Ciprofloxacin auf zehn Jahre erhöht werden (einzelne Chargen waren bis zu 13 Jahre haltbar).
Daten sind nicht in Stein gemeißelt!
Verfalldaten, Haltbarkeiten und Stabilitäten gebrauchsfertiger Zubereitungen sind eben nicht in Stein gemeißelt – sie können sich fortwährend unter Berücksichtigung aktueller Studienergebnisse, Stabilitätsuntersuchungen der Hersteller (on-going-stability) oder universitärer/institutioneller Arbeitsgruppen ändern.
Deshalb ist es legitim, im Rahmen einer Risikoabschätzung und Benutzung von Stabilitätsstudien eine erweiterte Haltbarkeit oder Stabilität festzulegen. Dies geschieht auf dem Boden wissenschaftlicher Tatsachen, und das Resultat muss dann vom jeweiligen Apotheker vertreten werden. Dafür hat er ein fundiertes wissenschaftliches Studium an einer Universität absolviert, welches ihn dazu befähigt!
Der Fall Bortezomib – und die finanziellen Folgen
In aller Munde – spätestens seit der Sendung „Panorama“ vom 1. September 2016 – ist der Proteasomen-Inhibitor Bortezomib (Velcade®). Der Hersteller Janssen-Cilag gibt in der aktuellen Fachinformation 100112793 (Januar 2016 [3]) unter „6.3 Dauer der Haltbarkeit“ Folgendes an:
„Die chemische und physikalische Stabilität der gebrauchsfertigen Lösung wurde für 8 Stunden bei 25 °C in der Originaldurchstechflasche und/oder einer Spritze belegt.“
Auf diese sehr kurze Stabilitätsaussage beruft sich auch die befragte Onkologin in der Panorama-Sendung mit der sinngemäßen Aussage, „die Lösung würde nicht mehr frisch aussehen“. Eine solche Aussage nach lediglich optischer/sensorischer Begutachtung zu treffen, ist ohne Zweifel diskussionswürdig.
Wie oben bereits angeführt, hat der pharmazeutische Unternehmer andere Interessen als z. B. der Krankenhausapotheker, welcher die Kostensituation im Auge behalten muss. Er ist nicht an langen Haltbarkeiten der gebrauchsfertigen Lösungen interessiert, denn diese mindern den Umsatz und damit die Zahl der abgesetzten Packungen. Interessanterweise werden diese verlängerten Haltbarkeiten gebrauchsfertiger Lösungen oder auch rekonstituierter Stammlösungen nach Patentablauf und dem Eintritt generischer Wettbewerber als Marketinginstrument genutzt.
Dazu vergleiche man nur einmal die Haltbarkeitsangaben der Fachinformationen von beispielsweise Carboplatin. Auf fachinfo.de sind elf Handelspräparate gelistet. Sämtliche Zubereitungen enthalten als pharmazeutische Bestandteile Carboplatin und Wasser – lediglich das Präparat Carboplatin-GRY® 10 mg/ml enthält noch D-Mannitol.
Die Haltbarkeiten der gebrauchsfertigen Lösungen reichen von acht Stunden beim Präparat Carboplatin Bendalis® 10 mg/ml (unabhängig von Trägerlösung und Lagertemperatur) bis zu 56 Tagen (!) beim Präparat Carboplatin Hospira® 10 mg/ml (gelöst in Glucose 5%, Lagerung bei 2 – 8 °C unter Lichtschutz, PVC-freie Beutel, Zielkonzentration der Lösung: 0,2 – 3,5 mg/ml).
Die Firma onkovis GmbH bietet gleich zwei Carboplatin-Konzentrate an: Carboplatin onkovis 10 mg/ml Infusionslösung mit einer Haltbarkeit von 72 Stunden und Carboplat onkovis 10 mg/ml Konzentrat zur Herstellung einer Infusionslösung mit einer Haltbarkeit von 28 Tagen (in Glucose 5% gelöst, keine Temperaturangabe zur Aufbewahrung) gemäß der jeweils aktuellen Fachinformation.
Es stellt sich dann schon die Frage, wie diese großen Unterschiede zustande kommen, wenn praktisch alle Präparate – bis auf eines – nur Carboplatin und Wasser enthalten.
Ein Vial Bortezomib (Velcade®) 3,5 mg kostet netto im Einkauf etwa 1305 € (Stand 09/2016) und hat nach Rekonstitution gemäß Hersteller eine Haltbarkeit von acht Stunden. Bortezomib wird in praktisch allen Mono- und Kombinationstherapien in einer Dosierung 1,3 mg/m2 angewendet (s.c. oder i.v.). Die meisten Patienten haben eine KOF von 1,5 bis 2,0 m2. Dies entspricht einer Dosis von 1,95 bis 2,60 mg.
Wenn nur ein Patient an diesem Tag behandelt wird, entsteht ein Verwurf von 1,55 bis 0,90 mg (entsprechend ca. 578 bis 335 €). Hier stellt sich die Frage, wer diesen Verwurf bezahlt bzw. die Kosten des Verwurfes trägt? Es war klar, dass hier nach Lösungen gesucht wurde. Viele Arbeiten und Studien zur Stabilität von gebrauchsfertigen Bortezomib-Lösungen wurden durchgeführt, um dieses Kostenproblem in den Griff zu bekommen.
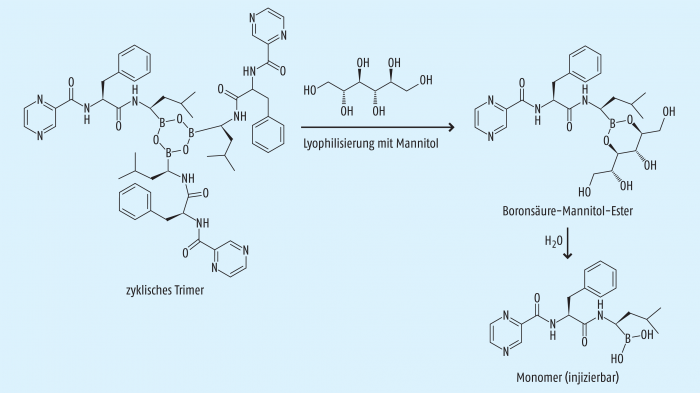
Bortezomib zählt zu den niedermolekularen Verbindungen (engl. small molecules). Es ist ein Boronsäure-Dipeptid mit den Aminosäuren Leucin und Phenylalanin. Die Stabilität von Bortezomib, welches streng genommen in drei molekularen Formen vorliegt (Abb. 1), ist so gut, dass das handelsübliche Lyophilisat bei Raumtemperatur gelagert werden kann.
Das Fertigarzneimittel enthält Bortezomib als Anhydrid und zyklisches Trimer (Boroxin-Derivat). Wenn es mit der zehnfachen Menge Mannitol lyophilisiert wird (ein Vial enthält 3,5 mg Bortezomib und 35 mg Mannitol), bildet sich ein Boronsäure-Mannitol-Ester, und im rekonstituierten Vial herrscht dann nach der teilweise erfolgten Hydrolyse ein Gleichgewicht zwischen dem Bortezomib-Monomer und dem Boronsäure-Mannitol-Ester (Abb. 1) [4].
Studien stützen längere Haltbarkeit
In einer NMR-Studie wurde der Abbau von Bortezomib-Lösungen unter klinischen Anwendungsbedingungen untersucht. Bortezomib wurde in steriler NaCl-Lösung 0,9% (in D2O = deuteriertes Wasser) rekonstituiert. Drei Muster wurden erzeugt:
- Vial A: ohne Vorbehandlung;
- Vial, B: der Luftraum im Vial wurde mit Argon gefüllt;
- Vial C: der Luftraum wurde mit Sauerstoff gefüllt.
Die Muster wurden bis zur Untersuchung bei 4 °C unter Lichtschutz gelagert. Begleitend zur NMR-Analytik wurde der Gehalt via HPLC kontrolliert. Nach einwöchiger Lagerung zeigte keines der drei Muster Abbau oder Zersetzung. Nach einem Monat konnten lediglich bei Muster C (Sauerstoff-Atmosphäre) kleine Mengen neugebildeter Produkte (2 Peaks) detektiert werden. Muster A und B (Argon-Atmosphäre) waren nach einem Monat unverändert [5].
Was sagt die Literatur?
Eine aktuelle Literaturrecherche via PubMed und www.stabilis.org erbrachte mehr als zehn verschiedene Stabilitätsstudien zu Bortezomib, welche unter Einsatz verschiedener Analysentechniken von unterschiedlichen Arbeitsgruppen erstellt wurden. Haltbarkeiten von bis zu 42 (!) Tagen [6] bei lichtgeschützter Lagerung im Kühlschrank (2 – 8 °C) lassen sich so abbilden.
Zwar sollten diese modifizierten Haltbarkeiten nicht bis zum Äußersten ausgereizt werden, aber eine risikobasierte verlängerte Haltbarkeit einer gebrauchsfertigen Bortezomib-Lösung von beispielsweise sieben Tagen bei lichtgeschützter Lagerung im Kühlschrank lässt sich bei dieser Fülle von Daten problemlos vertreten.
Wie eingangs bereits erwähnt, steht hier der jeweilige Hersteller – Apotheke oder Herstellbetrieb – für diese verlängerte Haltbarkeit gerade. Nicht anders verhält es sich mit den Daten der gerne verwendeten STABIL-Liste der ADKA [7].
Haltbarkeit von monoklonalen Antikörpern
Ein solches Prozedere funktioniert nicht nur bei den small molecules. Auch bei den schon länger im Markt befindlichen hochkomplexen monoklonalen Antikörpern (MABs) hat sich mittlerweile ein große Menge an Stabilitätsdaten angesammelt.
Trastuzumab (Herceptin®) ist laut deutscher Fachinformation (Stand: Februar 2016) nach Rekonstitution für 48 Stunden bei 2 bis 8 °C haltbar. Auf der Website der FDA (www.fda.gov) lässt sich ein Dokument über Trastuzumab abrufen, welches für die rekonstituierte Stammlösung eine Haltbarkeit von 28 Tagen bei 2 bis 8°C vorgibt (Webcode U6VE3). Dies ist immerhin ein Unterschied von 26 Tagen!
Geben Sie den Webcode U6VE3 auf www.deutsche-apotheker-zeitung.de in das Suchfeld ein und Sie gelangen direkt auf das FDA-Dokument.
Die anwendungsfertige Infusionslösung ist laut deutscher Fachinformation haltbar für 24 Stunden bei bis zu 30 °C. Eine Richtkonzentration (wie bei Rituximab) wird nicht empfohlen. Das berechnete Volumen an NaCl-Lösung 0,9% wird aus dem Infusionsbeutel entnommen und durch Herceptin-Stammlösung (21 mg/ml) ersetzt.
Beispiel: Errechnete individuelle Patientendosis = 420 mg = 20 ml Herceptin-Stammlösung. Es werden 20 ml NaCl 0,9% aus dem Beutel entnommen und durch 20 ml Herceptin-Stammlösung ersetzt.
Zwei Literaturstellen geben für Trastuzumab-Lösungen mit den Endkonzentrationen 0,8 mg/ml und 2,4 mg/ml eine Haltbarkeit von 180 Tagen (!) bei 4 °C an [8, 9]. Weitere Quellen geben für andere Endkonzentrationen 28 Tage bei Lagerung im Kühlschrank oder bei Raumtemperatur an [10].
MABs sind allerdings empfindlich bezüglich Lagerung und Transport. Die Einhaltung der Kühlkette, die Vermeidung von mechanischem und thermischem Stress, Lichtschutz und im Idealfall das Entlüften der Beutel (Luftsauerstoff) unterstützen die längeren Haltbarkeiten deutlich, denn bei den MABs können eine Vielzahl von Abbaureaktionen ablaufen:
- Denaturierung durch Temperatur und Scherkräfte,
- oxidative Abbaureaktionen,
- durch Licht vermittelte Abbaureaktionen.
Die durch thermischen, mechanischen oder chemischen Stress vermittelte Aggregatbildung führt in der Regel zu einem deutlichen Verlust der biologischen Aktivität und unter Umständen sogar zu ernsthaften immunogenen Reaktionen [11].
Vorsicht bei Neueinführungen
An ihre Grenzen stoßen die bisher dargelegten Punkte zu verlängerten Haltbarkeiten natürlich bei Neueinführungen oder relativ neuen Präparaten wie z. B. Nivolumab (Opdivo®). Hier werden sicherlich noch Monate bis Jahre vergehen, bis erweiterte Stabilitätsdaten zur Verfügung stehen. Bis zu diesem Zeitpunkt sind natürlich die Angaben der Fachinformation bindend.
Bei den neuartigen Konjugat-Antikörpern wie z. B. Trastuzumab-Emtansin (Kadcyla®) ist noch nicht sicher zu beurteilen, ob der Linker, also das Bindestück zwischen Trastuzumab und Emtansin (DM1), über längere Zeit stabil ist (sowohl bei der Stammlösung als auch bei der anwendungsfertigen Infusionslösung).
Fazit
Pharmazie ist und bleibt eine Wissenschaft: Neue Erkenntnisse zur Stabilität von Arzneimitteln sollen und müssen (auch aus volkswirtschaftlichen Gründen) genutzt werden. Hier kann sich der Apotheker und Pharmazeut auch als Fachmann profilieren und dem „Schubladenzieher“ eine lange Nase zeigen.
Und damit ganz zum Schluss keine Missverständnisse aufkommen:
Ganz oben stehen auch bei der Anwendung neuer wissenschaftlicher Erkenntnisse die pharmazeutische Qualität und die Patientensicherheit! |
Quellen
[1] Lyon RC et al. J Pharm Sci 2006;95(7):1549-1560
[2] Brooke C et al. Biosecurity Bioterrorism 2009;7(1):101-107
[3] www.fachinfo.de/suche/fi/008387
[4] www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_chemr.pdf
[5] Bolognese A et al. An NMR Study of the Bortezomib Degradation under Clinical Use Conditions. Adv Hematol 2009, Article ID 704928
[6] Walker SE et al. Can J Hospital Pharm 2008;61(1):14-20
[7] STABIL-Liste, Irene Krämer, Judith Thiesen, ADKA
[8] www.stabilis.org/FichesBiblio/3343.pdf
[9] Paul M et al. Int J Pharm 2013;448(1):101-104
[10] Pabari R.M et al. Current Pharm Biotechnol 2013;14(2):220-225
[11] Garcia-Fruitos E et al. Current Pharm Biotechnol 2011;12(10):1530-1536
Autor
Dr. Christian Beck
1995: Pharmaziestudium an der Goethe-Universität, Frankfurt/Main;
2006: Dr. phil nat. in Pharmazeutischer Biologie an der Universität Frankfurt/Main;
2007 - 2009: Krankenhausapotheker, Leiter der Qualitätskontrolle und Qualified Person in der Uniklinik, Frankfurt/Main;
2009 - 2012: Auditor und Qualified Person bei Fresenius Kabi Deutschland;
2012 - 2013: Qualified Person § 14 AMG & Leiter der Herstellung bei Aukamm Pharma GmbH & Co. KG, Wiesbaden
2013 - 2016: Apotheker in der Aukamm-Apotheke Sterilproduktion, Wiesbaden;
seit 2016: Qualified Person Leiter der Qualitätskontrolle bei Rhein Main Compounding GmbH
Lesen Sie hierzu auch:
- "Niemand weiß, was beim Transport geschieht!" - Klinikapotheker warnt vor unkritischem Gebrauch von Stabilitätsdaten
- Rechtlich verbindlich? Was gilt bei unterschiedlichen Angaben zur Haltbarkeit von Zytostatika-Zubereitungen?
- Warum macht die AOK das? Wie Kassen die exklusiven Zyto-Verträge rechtfertigen



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.