

















- DAZ.online
- News
- Debatte & Meinung
- Was ist eine Rezeptur...
Kommentar
Was ist eine Rezeptur? – Klären statt Abwarten
Süsel - 12.08.2021, 12:15 Uhr

DAZ-Redakteur Dr. Thomas Müller-Bohn fordert eine grundlegende Klarstellung, wie Rezepturen in all ihren Varianten zu definieren sind. (Foto: Victor Moussa /stock.adobe.com)
Die jüngste Entwicklung um die Opiumtinktur ist aus berufspolitischer Sicht ein weiterer Aspekt der seit Jahrzehnten geführten Verfahren um das Rezepturprivileg. Den Zusammenhang des aktuellen Falls mit dieser langfristigen Entwicklung beschreibt DAZ-Redakteur Dr. Thomas Müller-Bohn in einem Kommentar.
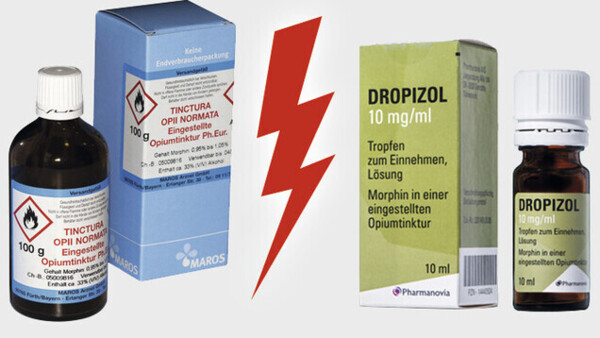
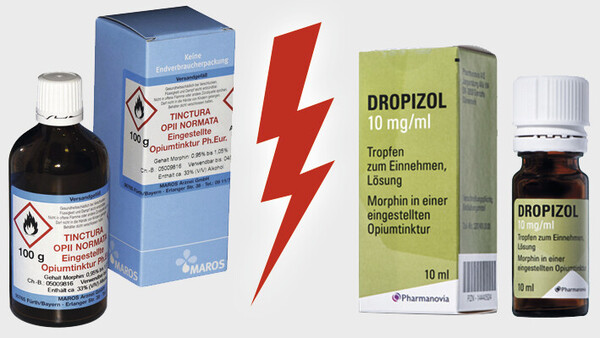
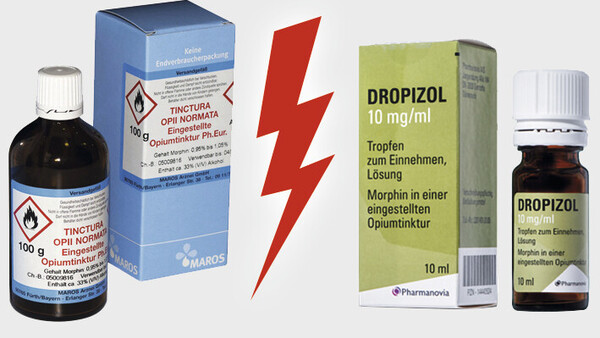
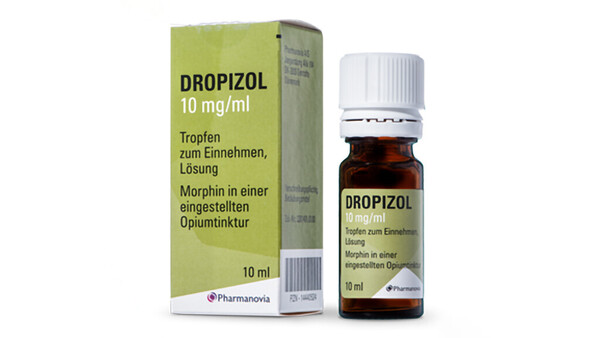
Ein erfundenes Szenario könnte nicht stärker überzeichnet werden als die Konstellation, die sich derzeit rund um die Opiumtinktur bietet. Sie enthält genug Stoff für mehrere Dramen. Besonders beeindruckend, dass es dabei um eines der ältesten noch angewendeten Arzneimittel geht.
Kaum ein anderes Produkt könnte den Archetypus eines Arzneimittels besser verkörpern, das in der Apotheke für einen Patienten individuell abgefüllt wird, gestützt auf die Qualitätsvorgaben des amtlichen Arzneibuchs. Das ist das klassische Dispensieren, wie es seit Jahrhunderten praktiziert wird, Apothekertätigkeit in Urform. Doch einige Gerichte wollen nun erkennen, dass dies keine Rezeptur wäre. Das Produkt soll angeblich ein Fertigarzneimittel sein, obwohl es aus einer Zeit stammt, in der es so etwas noch gar nicht gab. Wie deutlich kann es noch werden, dass an diesen Auslegungen irgendetwas nicht folgerichtig sein kann?
Mehr zum Thema

Zukunft des Rezepturprivilegs
Von der Opiumtinktur zur allgemeinen Sorge um die Rezeptur
Es wird allerhöchste Zeit, diese Argumente zu hinterfragen und dabei auch den Versorgungszweck der Rezeptur zu bedenken. In der seit Jahrzehnten andauernden Serie von Urteilen, die das Defektur- und Rezepturprivileg einengen, ging es meist um ausgefallene Produkte, die nur in wenigen Apotheken interessierten. Darum gab es bisher keinen Aufschrei. Doch nun betrifft es ein Produkt mit mehr Versorgungsrelevanz, das in vielen Apotheken zumindest gelegentlich vorkommt.
Darum sorgen sich nun viele Apotheker. Wenn ein Produkt fällt, könnte auch das Abfüllen anderer Arzneimittel infrage gestellt werden. Die politisch aufmerksam verfolgte Versorgung mit Cannabisblüten stellt praktisch dieselbe Konstellation dar. Wenn dann noch weitere Ecken herausbrechen, bliebe bald nicht mehr genug übrig, um die Rezeptur als Institution zu erhalten.
Katastrophe für Apotheker und Patienten
Das wäre eine Katastrophe für das Selbstverständnis und die Außendarstellung der Apotheker und ein willkommenes Argument für die Versender. Denn dann entfiele ein wesentlicher Unterschied zwischen Vor-Ort-Apotheken und Versand. Doch vor allem wäre es eine Katastrophe für die Patienten. Denn ein wichtiger Teil der Versorgung stünde nicht mehr zur Verfügung und für Krisenzeiten wäre ein entscheidendes Stück Sicherheit verloren. Die Erinnerung an die Herstellung von Desinfektionsmitteln ist noch frisch. Dies alles spricht dafür, das Thema nun endlich vom Ende her zu denken.
Ein Fall, viele Dramen
Doch im Fall der Opiumtinktur sind noch mehr Geschichten zu erzählen. Geradezu klischeehaft stehen sich ein Kleinunternehmen und ein Anbieter im Besitz eines internationalen Private-Equity-Fonds gegenüber. Wenn den Apotheken das Abfüllen der Tinktur verboten würde, wäre der Hauptlieferant des Produkts in seiner Existenz bedroht. Ein weiterer Aspekt betrifft die Produkte. Es geht es um wirkstoffgleiche Arzneimittel mit einem erheblichen Preisunterschied, wobei das teurere Fertigarzneimittel letztlich von generationenlangen Erfahrungen mit dem abgefüllten Rezepturprodukt inspiriert wurde. Mit dem finanziellen Aspekt dürften auch die Krankenkassen zumindest als aufmerksame Beobachter angesprochen sein.
Apotheker zwischen den Fronten
Zwischen allen diesen Fronten stehen die Apotheken. Der aktuelle Streit zwischen den Herstellern vermischt sich mit anderen Angriffen auf die Rezeptur. Wenn die Apotheker an einer Stelle zurückweichen, könnte ihre ganze Position zu diesem Themenkreis in Gefahr geraten. Das bringt die Apotheken in ein Dilemma: Wer sich in juristischer Hinsicht gerne vorsichtig verhalten möchte, kann damit seine berufspolitische Position gefährden. Die juristische Vorgehensweise ist von der politischen Position nicht mehr zu trennen. Nun rächt sich, dass das Thema so lange vernachlässigt wurde.
Es bleibt die Hoffnung auf eine schnelle Klärung. Diese ist aufgrund der Verfahrenswege weder juristisch noch politisch zu erwarten. Doch zumindest für den Fall der Opiumtinktur kann das Bundesinstitut für Arzneimittel und Medizinprodukte als zuständige Bundesoberbehörde über die Zulassungspflicht entscheiden. Dies könnte den aktuellen Fall vermutlich beenden. Doch falls die Behörde die Zulassungspflicht negiert, sollten die Apotheker sich nicht zurücklehnen, sondern die derzeitige Entwicklung als Warnung nehmen. Das Thema lässt sich durch Abwarten nicht bewältigen. Die juristischen Grundfragen zur Definition von Rezepturen in allen ihren Varianten müssen rechtssicher beantwortet werden – im Interesse der Apotheken und vor allem der Patienten.



9 Kommentare
Zulasssung ist nicht DER Problemlöser
von norbert brand am 17.08.2021 um 9:49 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Ist es so eindeutig?
von Michael Mischer am 16.08.2021 um 9:46 Uhr
» Auf diesen Kommentar antworten | 4 Antworten
AW: Zwar im Gesetz versteckt, aber dennoch eindeutig!
von Andreas P. Schenkel am 22.08.2021 um 20:16 Uhr
AW: Ist es so eindeutig
von Michael Mischer am 22.08.2021 um 21:46 Uhr
AW: Normal-neue Arzneimittel
von Andreas P. Schenkel am 23.08.2021 um 0:05 Uhr
AW: Ist es so eindeutig
von Michael Mischer am 23.08.2021 um 10:24 Uhr
"Umgehung" der Zulassung?
von norbert brand am 13.08.2021 um 9:31 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Weitere Aspekte des Dillemmas
von Michael Mischer am 13.08.2021 um 8:14 Uhr
» Auf diesen Kommentar antworten | 1 Antwort
AW: Da purzelt aber einiges durcheinander!
von Andreas P. Schenkel am 14.08.2021 um 18:17 Uhr
Das Kommentieren ist aktuell nicht möglich.