












- DAZ.online
- DAZ / AZ
- DAZ 15/2021
- Woher kommt SARS-CoV-2
Pandemie Spezial
Woher kommt SARS-CoV-2?
Schon Wochen vor dem ersten dokumentierten Fall muss das Virus auf Menschen übergesprungen sein
Da nach der Meldung der ersten Fälle die Zahl der Patienten innerhalb kurzer Zeit exponenziell stieg, wurde die Provinz Hubei mit einem halben Dutzend Städten von mehreren Millionen Einwohnern am 23. Januar 2020 durch einen drakonischen cordon sanitaire von der Außenwelt abgeschlossen. Wie man heute weiß zu spät, denn zwischenzeitlich war SARS-CoV-2 durch Reisende mit von Wuhan ausgehenden Direktflügen bereits nach Taiwan (erster dokumentierter Fall am 21. Januar 2020) sowie nach Frankreich und Deutschland (24. Januar 2020 bzw. 27. Januar 2020) gelangt. Von den offiziell bis zum 21. Februar 2020 in Europa dokumentierten Fällen traten 14 Erkrankungen bei Reisenden auf, die direkt aus China kamen, 14 standen im Zusammenhang mit dem Ausbruch in Stockdorf in Bayern, der wiederum durch eine Reisende aus China verursacht worden war. Für zwölf Erkrankungen blieb der Ursprung der Infektion unbekannt.
Spätestens seit Sommer 2020 ist bekannt, dass die offizielle Chronologie nicht den Tatsachen entspricht (siehe Feldmeier H. Eine Spurensuche – Molekulare Epidemiologie hilft, die verschlungenen Wege von SARS-CoV-2 zu entschlüsseln. DAZ 2020, Nr. 26, S. 22 – 24). Eine retrospektive Analyse von im Großraum Paris gesammelten Blutproben zeigte, dass in Frankreich bereits im November 2019 Menschen mit SARS-CoV-2 infiziert waren [1]. Diese Beobachtung wird durch Daten aus Italien [2] und der Schweiz [3] unterstützt. Aber wie kann es sein, dass das Virus bereits im November in mehreren europäischen Ländern – wenn auch unbemerkt – zirkulierte, wenn der erste von den chinesischen Behörden bestätigte Krankheitsfall auf die letzten Dezembertage datiert wird?
Virologische Detektivinstrumente
Tritt erstmals ein neuartiges Virus auf, lässt sich über den Verwandtschaftsgrad von aus Patienten isolierten Genomen, dem Zeitpunkt, an dem das Virus aus einem Menschen isoliert wurde, und dem Ort, an dem die Infektion stattgefunden hat, auf den letzten gemeinsamen Vorfahren zurückschließen. Voraussetzung ist allerdings, dass möglichst viele zu unterschiedlichen Zeitpunkten an verschiedenen Orten gewonnene Proben vorliegen und die Mutationsrate des Erregers bekannt ist. Für SARS-CoV-2 wird die Mutationsrate mit 1,1 bis 1,3 × 10–3 Basensubstitutionen pro site pro Jahr angenommen [11].
Können die isolierten Genome einem geografischen Ort zugeordnet werden, lässt sich auch die „Richtung“ erkennen, in die sich das Virus bewegt hat. So konnte der Düsseldorfer Genomforscher Alexander Dilthey zeigen, dass im März 2020, SARS-CoV-2 die Landeshauptstadt nicht vom Hotspot Heinsberg aus erreichte (wie bislang vermutet wurde), sondern bereits vorher Virusgenome aus mehreren Kontinenten in der Stadt zirkulierten.
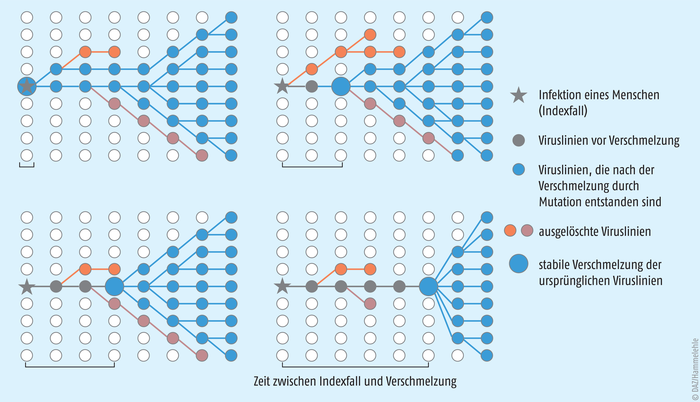
Abb.: Vier Szenarien der Ausbreitungsdynamik eines neuartigen zoonotischen Virus. Oben links: die Verschmelzung der ursprünglichen Viruslinien zu einer neuen Virusspezies erfolgt gleichzeitig mit dem Indexfall. Es kommt rasch zu einer epidemischen Ausbreitung. Oben rechts: die Verschmelzung erfolgt leicht zeitversetzt. Unten links: die Verschmelzung erfolgt etwas später. Die epidemische Ausbreitung erfolgt dementsprechend später. Unten rechts: die Verschmelzung erfolgt Wochen bis Monate nach dem Indexfall. Die Epidemie wird erst zeitverzögert erkannt, wie es die Modellrechnung für den Ursprung von SARS-CoV-2 nahelegt (nach [4]).
Spurensuche mit phylogenetischen Analysen
Dieser Frage ist eine Gruppe von Wissenschaftlern um den Evolutionsbiologen Joel Wertheim von der University of California in San Diego nachgegangen. Die Studie wurde gerade im Fachjournal „Science“ veröffentlicht [4]. Die bisher durchgeführten rückwärts gerichteten phylogenetischen Analysen, in denen mithilfe einer molekularen Uhr versucht wurde, auf das sogenannte Letzter-gemeinsamer-Vorfahr-Ereignis zurückzurechnen – also dem Zeitpunkt, an dem das Virus erstmalig einen Menschen infizierte –, haben sich als unzuverlässig erwiesen [5, 6]. Daher entwickelten die amerikanischen Wissenschaftler eine neue Methodik, um den Zeitpunkt des ersten Auftretens von SARS-CoV-2 in China einzukreisen. Sie verbesserten das genetische Verfahren und kombinierten es mit einem vorwärts gerichteten, hoch-komplexen biostatistischen Modell zur Ausbreitungsdynamik von SARS-CoV-2. In einem zweiten Schritt überprüften sie die Robustheit ihrer Methodik, in dem sie die Eingangsparameter des Modells solange modifizierten, bis die statistischen Aussagen über den Zeitpunkt des ersten Falls in einem engen Vertrauensbereich blieben. Als Datenbasis nutzten sie 583 SARS-CoV-2-Genome, die von Patienten in China zwischen Dezember 2019 und April 2020 isoliert worden waren.
Das Ergebnis der Modellierung ist frappierend: Die Elf-Millionen-Stadt Wuhan ist nicht der Ort, an dem SARS-CoV-2 von einem Tier auf einen Menschen übersprang. Das Virus muss bereits sechs bis zehn Wochen vor dem ersten dokumentierten Fall in Wuhan mehrfach von Mensch zu Mensch übertragen worden sein. Vermutlich waren die Infektionsketten aber sehr kurz. Der früheste Zeitpunkt der Erstinfektion ist nach der Modellrechnung Mitte Oktober 2019 und der späteste Mitte November 2019.
Damit ist die Hypothese, dass der Fisch- und Wildtiermarkt in Wuhan der Auslöser der Pandemie sei, vom Tisch. Vielmehr war der Húanan-Markt nur das allererste ausbruchsfördernde Ereignis, bei dem das neuartige Virus seine enorme Infektiosität und sein außerordentliches krankmachendes Potenzial bewies. Aus heutiger Sicht war der Markt in Wuhan nur die Lunte, die die Pandemie zündete. Zwischen Mitte Oktober und Mitte November 2019 hatten sich bereits diverse Lunten entzündet, die aber offensichtlich „ausbrannten“, bevor es zu einem Superspreading-Event kam. Wie von anderen zoonotischen Viren (beispielsweise dem Ebola-Virus) bekannt, musste SARS-CoV-2 in eine Stadt gelangen, in der viele Menschen regelmäßig engen Kontakt haben, um eine Epidemie auszulösen. Anderenfalls wäre es nach einigen wenigen Übertragungen wieder verschwunden.
Die Berechnungen der amerikanischen Evolutionsbiologen unterstützen auch die Hypothese, dass sich SARS-CoV-2 ursprünglich in einer Fledermaus entwickelt hatte (vermutlich aus der Familie der Hufeisennasen, Rhinolophus ssp.), dann entweder direkt oder indirekt über ein Schuppentier (Pangolin) auf einen Menschen übersprang [7]. Zu dieser Schlussfolgerung kommt auch die WHO-Expertengruppe, die sich zwei Wochen in China aufgehalten und gerade ihren Bericht veröffentlicht hat [8]. Ob das Überspringen in der Provinz Hubei stattfand oder – wie andere Wissenschaftler meinen – in der weiter südlich gelegenen Provinz Guandong [9], würde sich nur dann klären lassen, wenn systematisch (z. B. von stationär behandelten Patienten) gewonnene archivierte Proben aus beiden Provinzen untersucht werden könnten. Sollte es diese Proben gegeben haben, so werden sie von den chinesischen Behörden entweder unter Verschluss gehalten oder sie wurden zwischenzeitlich vernichtet.
Ein Nebenbefund der mathematischen Modellrechnungen ist, dass etwa zwei Drittel der Corona-Virus-Linien, die den Artensprung schafften, ausstarben, bevor die Infektionsketten sich verzweigen konnten. Laut Joel Wertheim gilt dieses Merkmal auch für andere zoonotische Viren [4] und ist ein Indiz dafür, wie viel Glück die Menschheit in den vergangenen hundert Jahren hatte, in denen sich die Zahl der durch zoonotische Viren verursachten Epidemien an zwei Händen abzählen lässt [4]. Die Studie der Evolutionsbiologen unterstreicht noch einmal die Notwendigkeit, endlich ein globales Netzwerk zur Überwachung viraler Zoonosen zu implementieren [10]. |
Literatur
[1] Carrat F et al. Evidence of early circulation of SARS-CoV-2 in France: findings from the population-based „CONSTANCES“ cohort. Eur J Epidemiol 2021;36:219-222, doi.org/10.1007/s10654-020-00716-2
[2] Apolone G et al. Unexpected detection of SARS-CoV-2 antibodies in the prepandemic period in Italy. Tumori Journal 2020, doi.org/10.1177/0300891620974755
[3] Jahn K et al. Detection of SARS-CoV-2 variants in Switzerland by genomic analysis of wastewater samples. medRxiv preprint 9. Januar 2021, doi.org/10.1101/2021.01.08.21249379
[4] Pekar J et al. Timing the SARS-CoV-2 index case in Hubei province. Science 2021, doi.org/10.1126/science.abf8003
[5] Mavian C et al. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. PNAS 2020, pnas.org/cgi/doi/10.1073/pnas.2007295117
[6] Zhang L et al. Origin and evolution of the 2019 novel coronavirus. Clin Infect Dis 2020;71(15):882-883, doi.org/10.1093/cid/ciaa112
[7] Li X et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol 2020;92(6):602-611, doi.org/10.1002/jmv.25731
[8] WHO-convened global study of origins of SARS-CoV-2: China Part; www.who.int/publications/i/item/who-convened-global-study-of-origins-of-sars-cov-2-china-part
[9] Lu J et al. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020;181:997-1003, doi.org/10.1016/j.cell.2020.04.023
[10] Mallapaty S. The search for animals harbouring coronavirus – and why it matters. Nature 2021;591:26-28, www.nature.com/articles/d41586-021-00531-z
[11] Martin M et al. Insights from SARS-CoV-2 sequences. Science 2021;371(6528):466-467, doi.org/10.1126/science.abf3995
Weitere Beiträge des Pandemie Spezials in DAZ 2021, Nr. 15
- Testangebot – die wirtschaftliche Seite: Kostenrechnung für Corona-Schnelltests
in Apotheken - Ein bisschen Christkindlesmarkt-Feeling: So testet die Marien Apotheke Bodenmais
- Die verwaiste Kosmetikkabine: So testet die Vital-Apotheke Bad Saulgau
- Zu zweit und nach dem Vier-Augen-Prinzip: So testet die Rathaus Apotheke Hagen
- Schnelltest-Zelt hält Wettertest nicht stand: So testet die Alte Apotheke Feuerbach – theoretisch
- Zwei Impfstoffe für die Arztpraxen: Neue Bestellregeln für Comirnaty® und Vaxzevria®
- Extra-Service ermöglicht Impfstoffanlieferung: Auch auf Amrum können die Hausärzte gegen Corona impfen – dank Apotheke und Großhandel
- Nach Verdünnung sechs Stunden haltbar? Rechtliche und pharmazeutische Aspekte rund um die Impfstoffaufbereitung
- Diabetes in Zeiten der Pandemie: Ein gut eingestellter Stoffwechsel schützt vor schweren COVID-19-Verläufen
- Das Dilemma der SARS-CoV-2-Impfstoffbeurteilung: Ein Gastkommentar
- Corona-Ticker: Neues zu SARS-CoV-2 in Kürze



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.